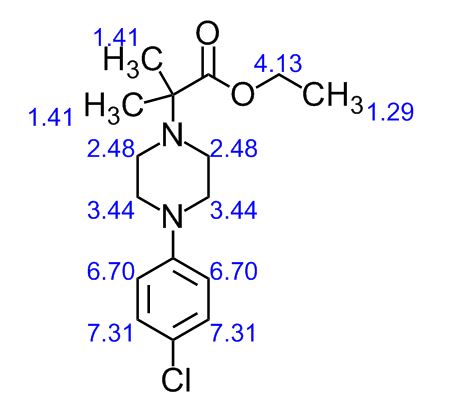
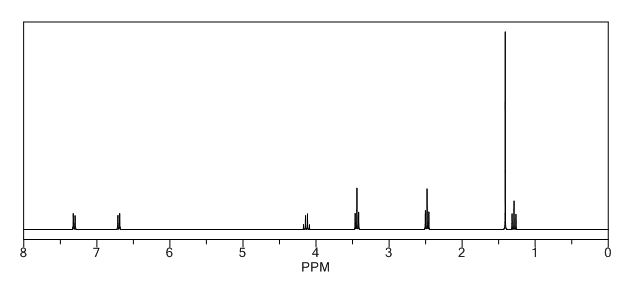
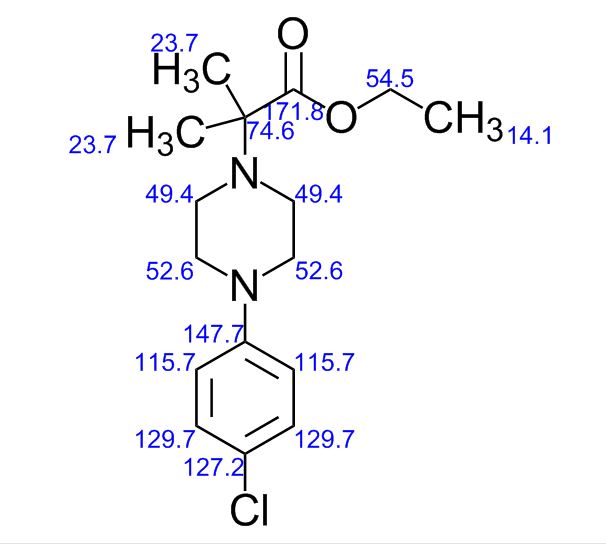
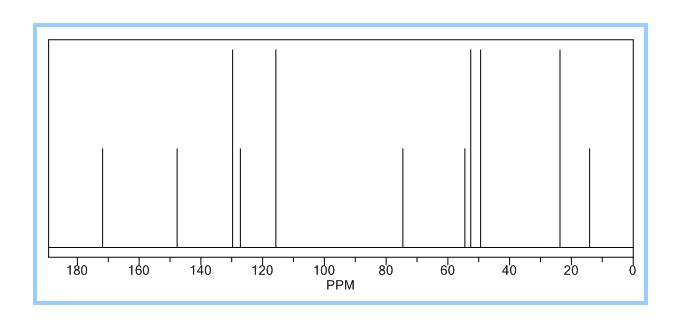
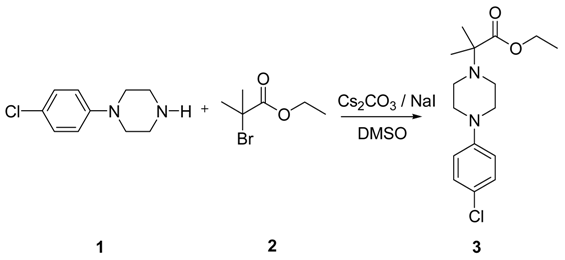
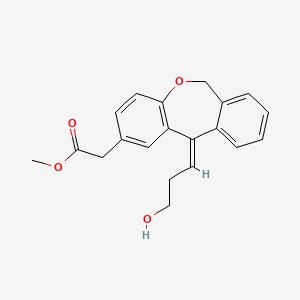
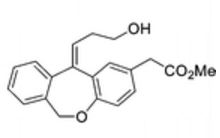
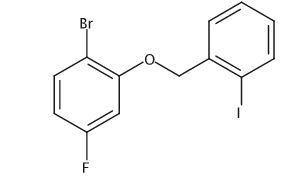
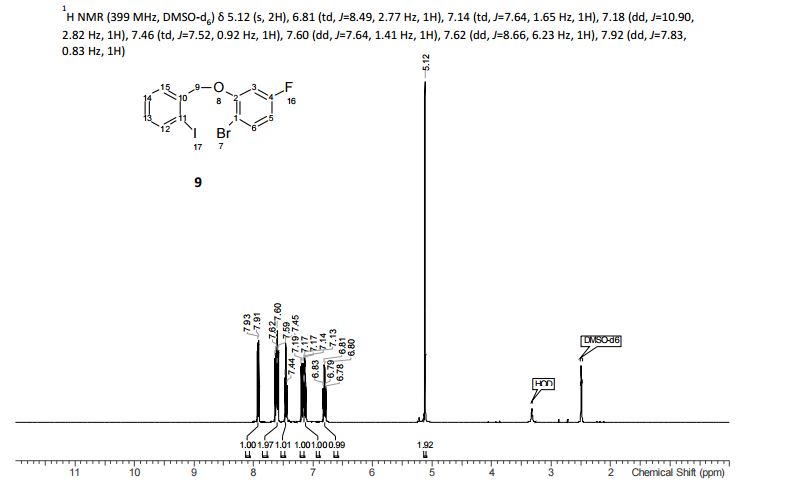
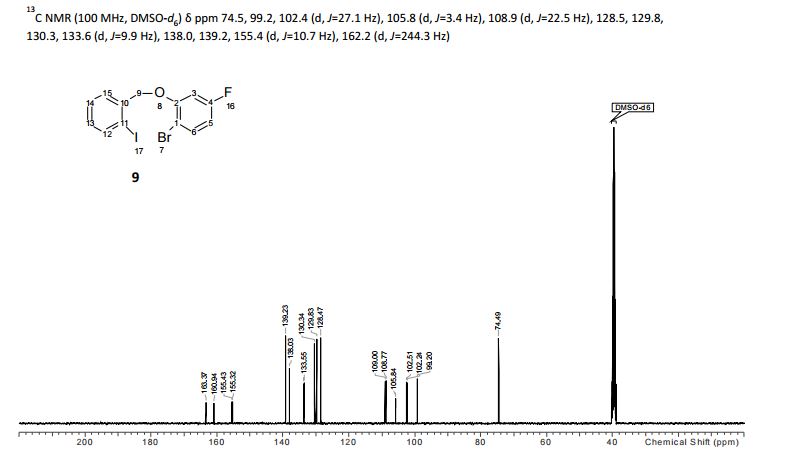
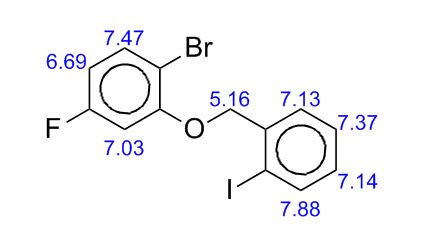
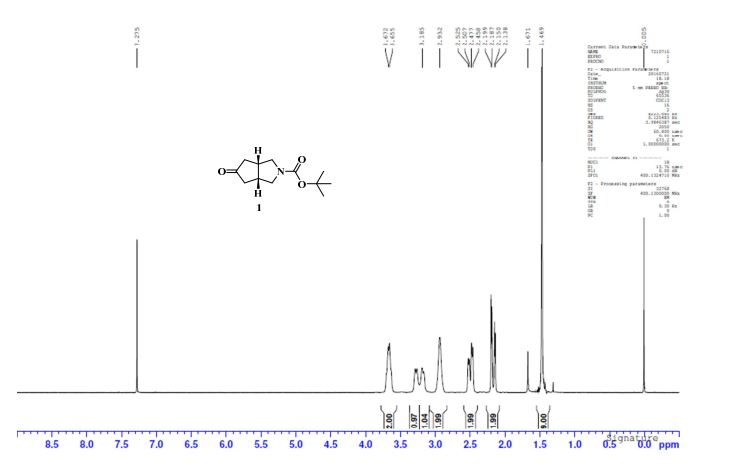
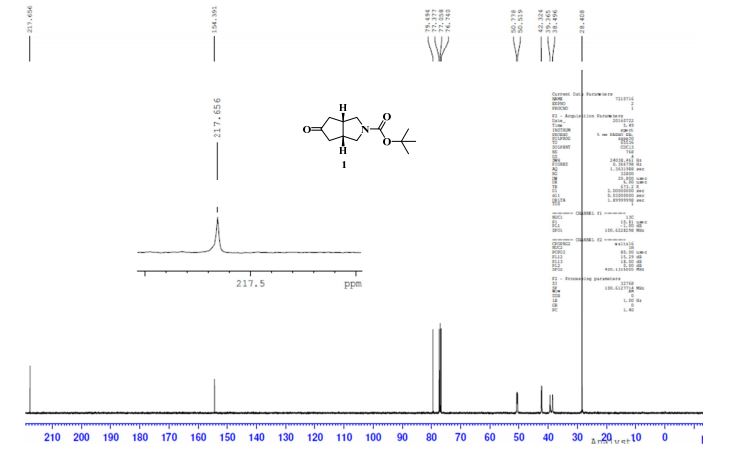
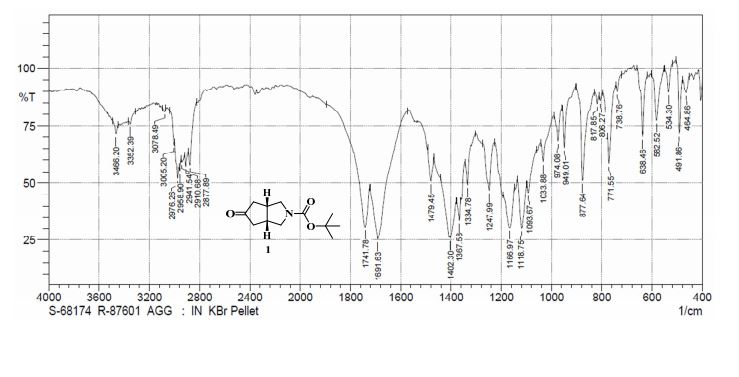
To a solution of 4-(4-chlorophenyl)piperazine dihydrochloride 1 (5.0 g, 0.0185 mol) in DMSO (30 ml), anhydrous cesium carbonate (30.0 g, 0.0925 mol), sodium iodide (1.39 g, 0.0093 mol) and ethyl 2-bromo-2-methylpropanoate 2 (3.97 g, 0.02 mol) were added. The resulting mixture was stirred at 25-30oC for 12 hours. The reaction mass was diluted with water (200 ml) and extracted with ethyl acetate (2 x 200 ml). The ethyl acetate layer was washed with water (2 x 100 ml), dried over anhydrous sodium sulfate (10.0 g) and concentrated under vacuum. The crude product thus obtained was purified by column chromatography (stationary phase silica gel 60-120 mesh; mobile phase 10% ethyl acetate in hexane). The title compound 3 was obtained as a white solid (4.73 g, 82 %).
1Department of Chemistry, Sambalpur University, JyotiVihar-768019, Orissa, India
2Institute of Chemical Technology (ICT), Matunga, Mumbai-400019, Maharashtra, India
*Author to whom correspondence should be addressed.
Received: 17 May 2009 / Accepted: 30 June 2009 / Published: 27 July 2009
Image may be NSFW. Clik here to view.
Bijay K Mishra
Professor at Sambalpur University, Chemistry Department
Abstract
The title compound was synthesized by N-alkylation of 4-(4-chlorophenyl)piperazine with ethyl 2-bromo-2-methylpropanoate and its IR, 1H NMR, 13C NMR and Mass spectroscopic data are reported.
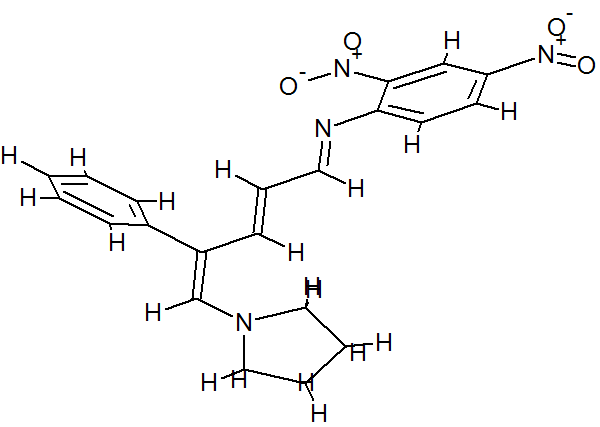
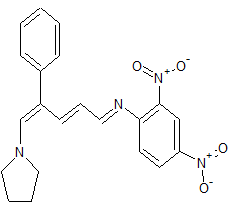
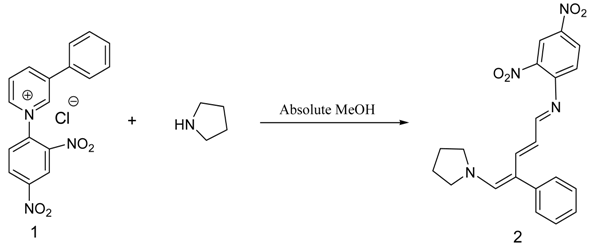
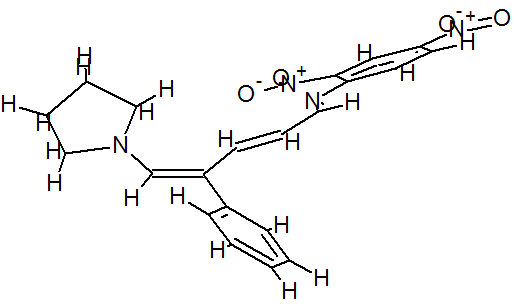
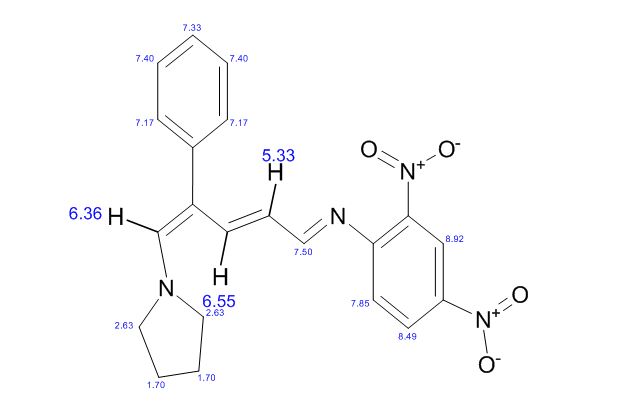
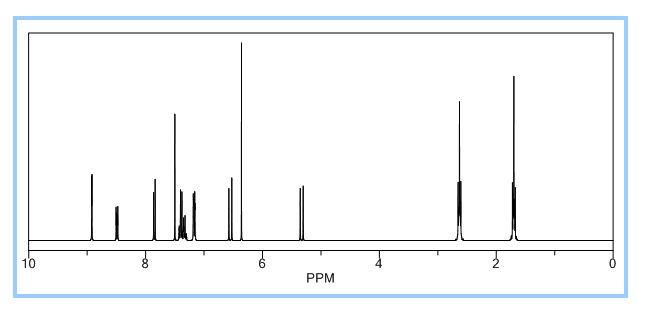
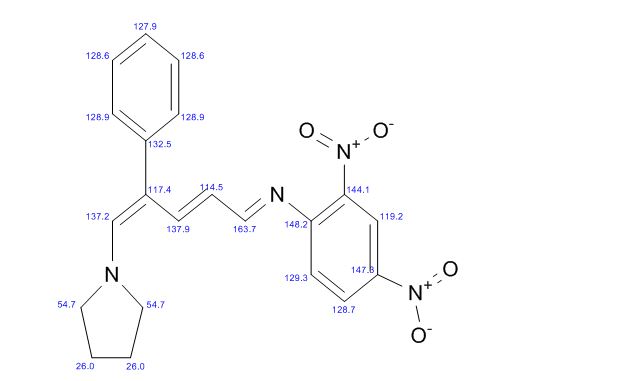
(E)-2,4-Dinitro-N-((2E,4E)-4-phenyl-5-(pyrrolidin-1-yl)penta-2,4-dienylidene) aniline dye was prepared in one pot by reaction of premade N-2,4-dinitrophenyl-3-phenylpyridinium chloride (DNPPC) and pyrrolidine in absolute MeOH.
Keywords:
N-2,4-dinitrophenyl-3-phenylpyridinium chloride (DNPPC); photochromic; pyridinium salt
N-2,4-Dinitrophenyl-3-phenylpyridinium chloride (DNPPC) 1 was prepared according to the literature method [1,2,3,4,5,6,7]. Recently, we became interested in the synthesis of photochromic compounds [8,9,10]. The UV-Vis spectra under irradiation of UV light of dye 2 indicate photochromic properties for this molecule. The salt 1 was premade and typically isolated and purified by recrystallization and characterized. To a solution of 1-chloro-2,4-dinitrobenzene (1.42 g, 7.01 mmol) in acetone (10 mL) was added 3-phenylpyridine (1.0 mL, 6.97 mmol). The reaction was heated at reflux for 48 h. The solvent was removed under reduced pressure and the red residue was stirred in hexanes. The precipitated product was collected by vacuum filtration to afford pure pyridinium salt 1 as a reddish brown solid (2.23 g, 6.25 mmol, 90%). 1H NMR (CDCl3, 500 MHz): δ (ppm) 9.9 (s, 1H), 9.4 (d, J = 6.0 Hz, 1H), 9.3 (d, J = 8.3 Hz, 1H), 9.2 (d, J = 2.2 Hz, 1H), 9.0 (dd, J = 8.7, 2.4 Hz, 1H), 8.5-8.6 (m, 2H), 8.0 (d, J = 7.3 Hz, 2H), 7.6- 7.7 (m, 3H); 13C NMR (CDCl3, 125 MHz): δ (ppm) 149.2, 145.6, 144.3, 144.2, 143.0, 139.2, 138.7, 132.5, 132.3, 130.6, 130.2, 129.6, 128.0, 127.6, 121.3; IR (KBr pellet) 3202, 3129, 2994, 2901, 1609 cm-1; m. p. = 182-183 °C; HRMS m/z Calcd for C17H12N3O4+ (M)+ 322.0828, found 322.0836.
Image may be NSFW. Clik here to view.
Reaction of pyrrolidine with salt (1) leads to the opening of the pyridinium ring and formation of dye 2. This dye was prepared from reaction of salt 1 (0.5 g, 1.4 mmol) in 5 mL absolute MeOH after cooling a reaction mixture to -10oC and keeping at this temperature for 15 min. To this was added pyrrolidine (0.1 g, 1.4 mmol) in 3 mL absolute MeOH over a period of 10 min. The prepared solid was filtered, washed with CH2Cl2, dried and recrystallized from n-hexane to yield 68% (0.37 g, 0.95 mmol) of pure metallic greenish-brown 2,
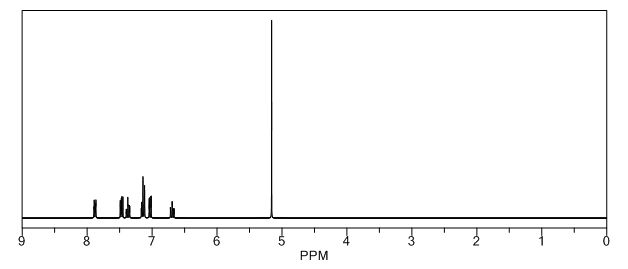
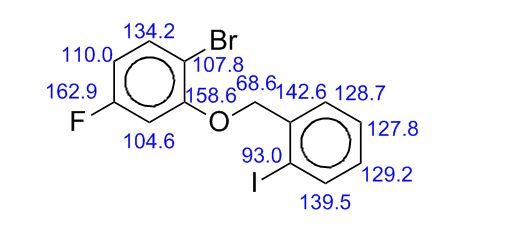
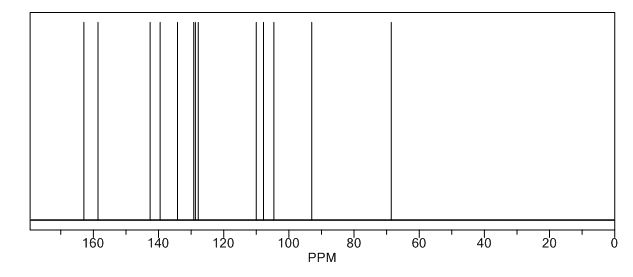
Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.
Image may be NSFW. Clik here to view. COSY PREDICT
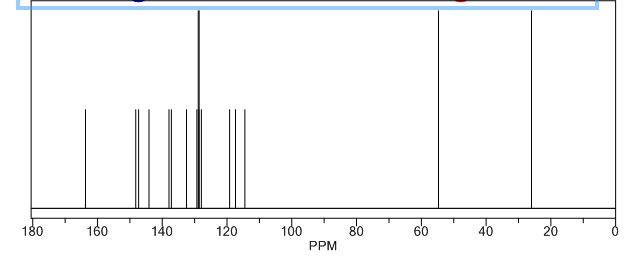
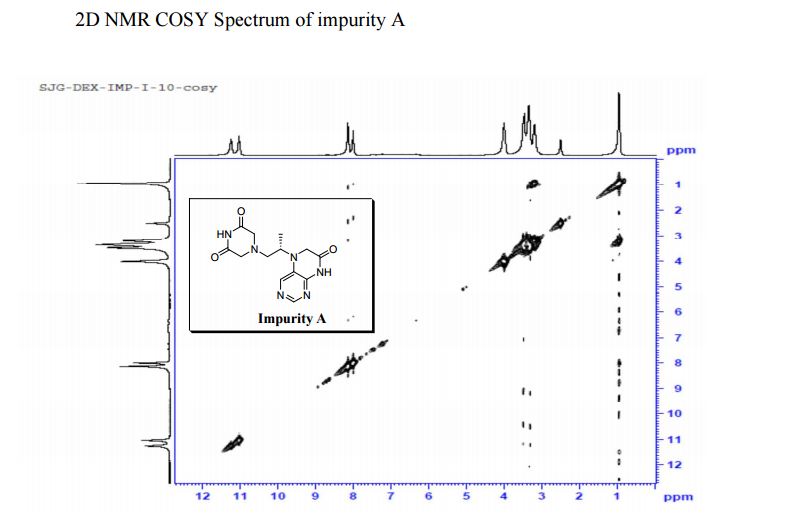
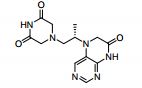
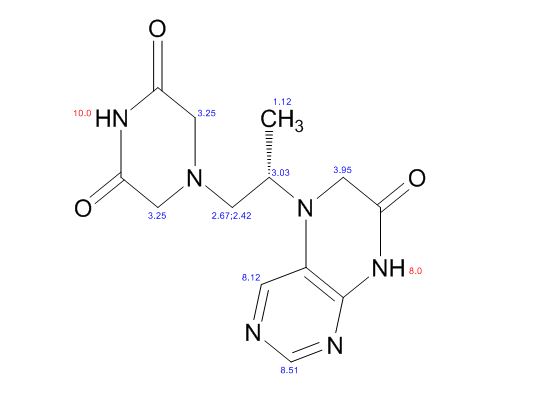
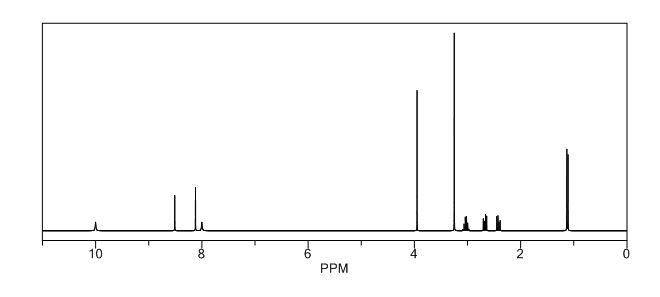
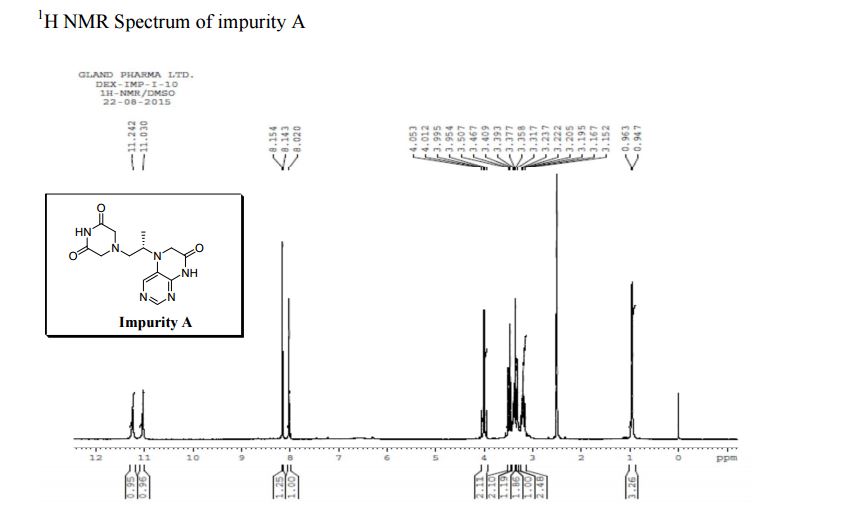
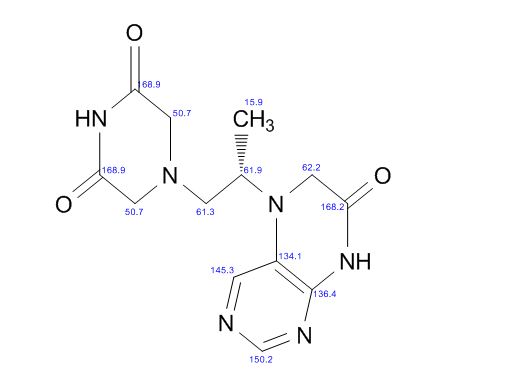
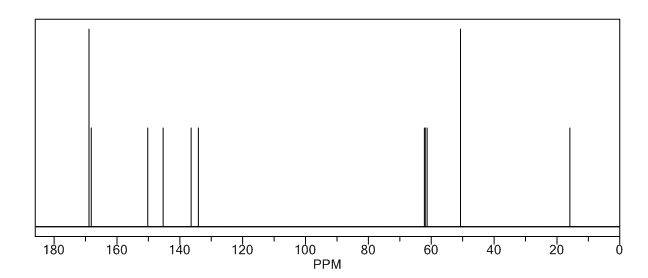
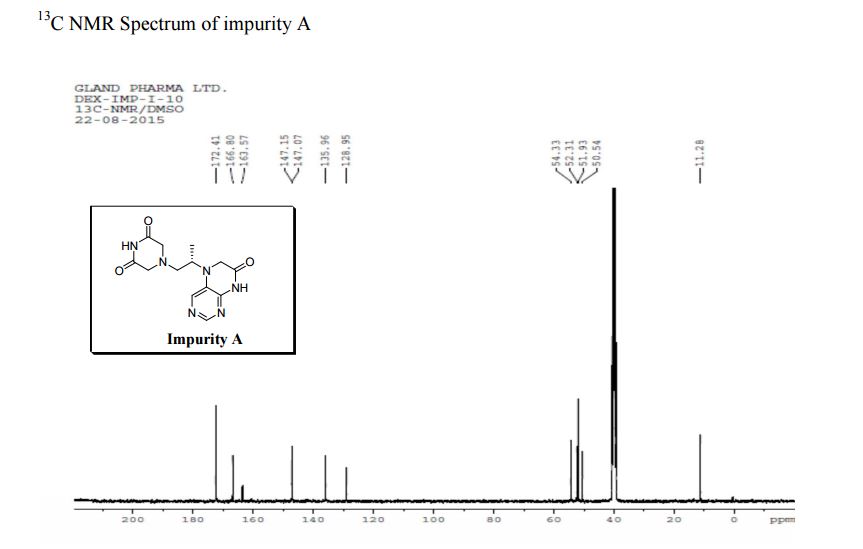
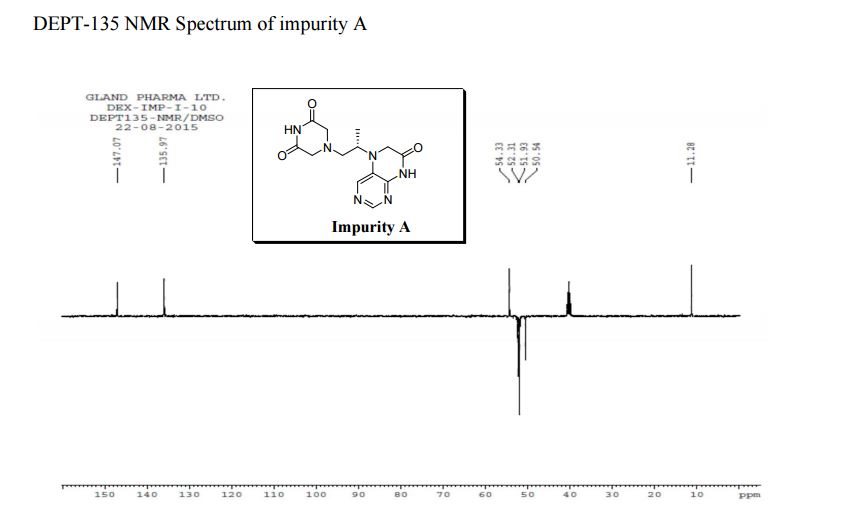
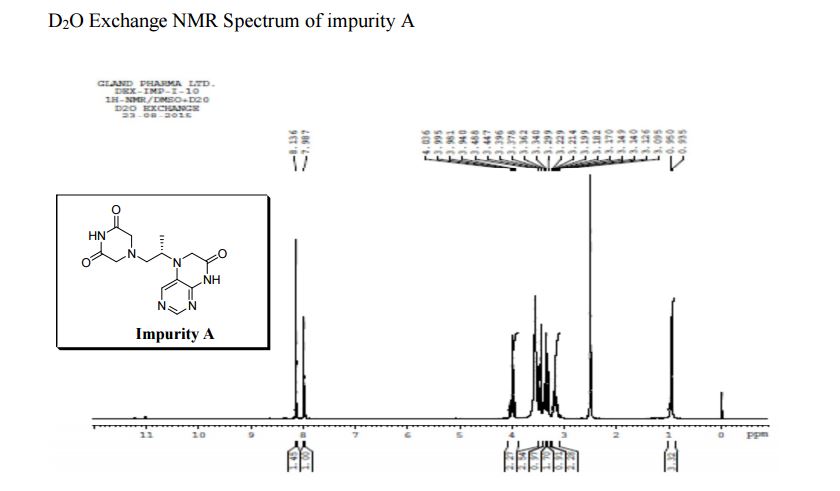
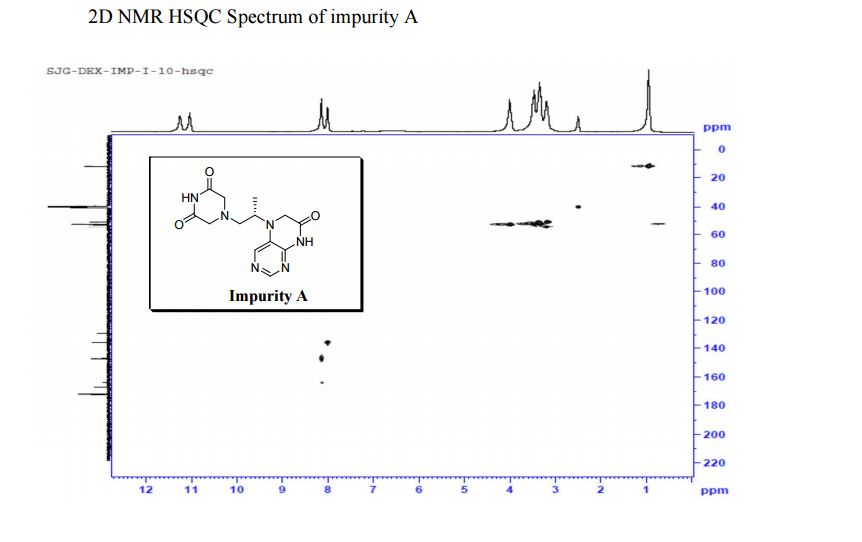
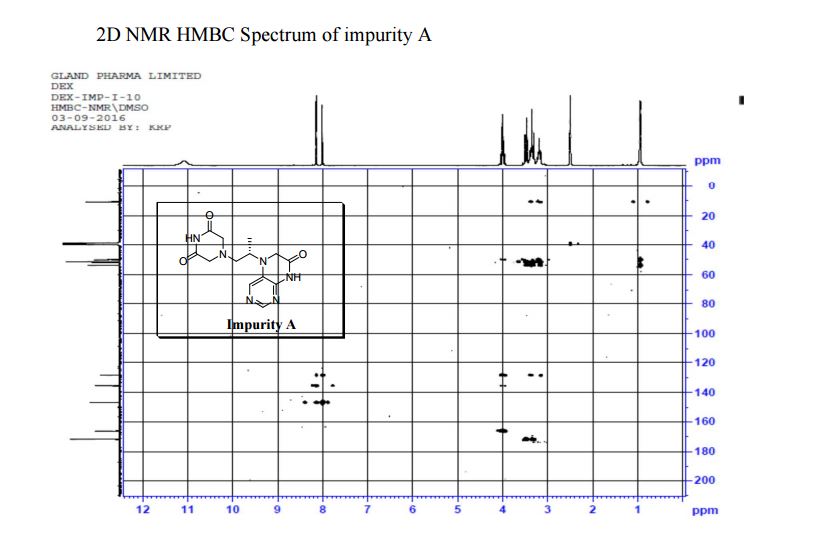
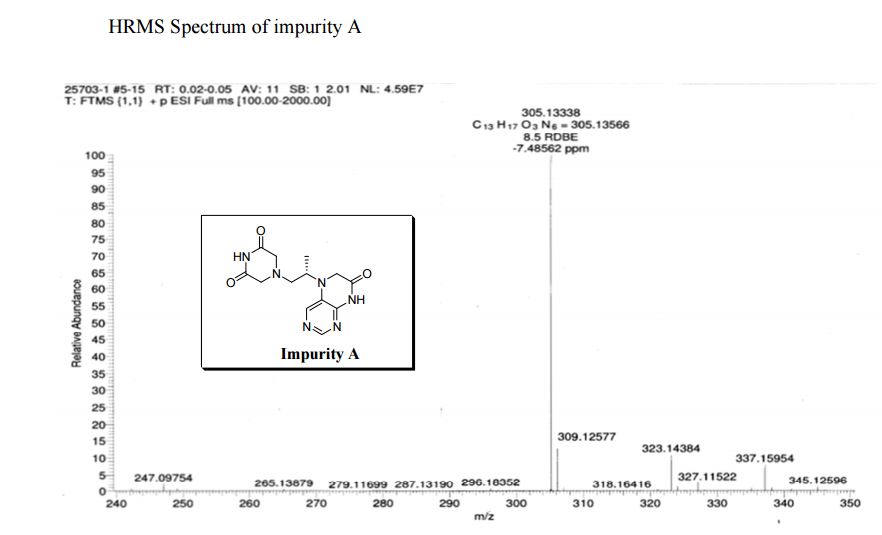
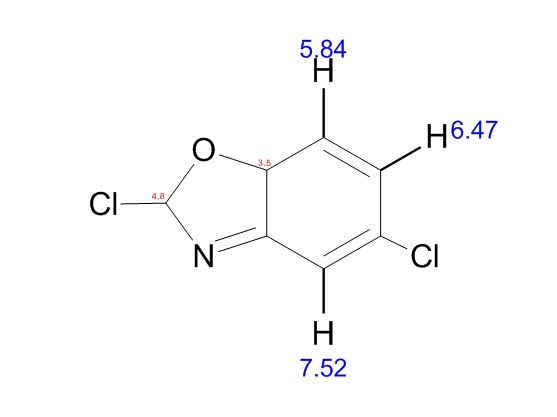
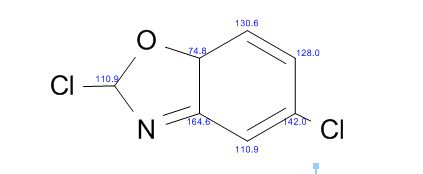
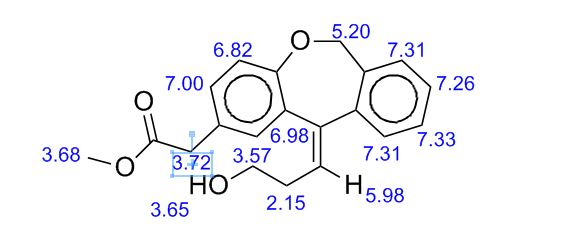
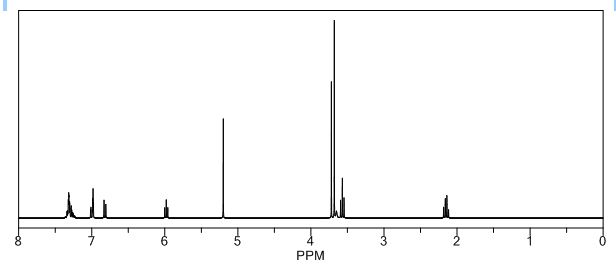
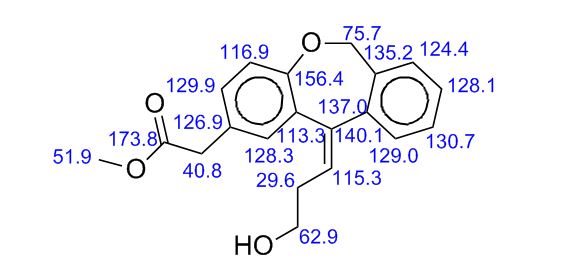
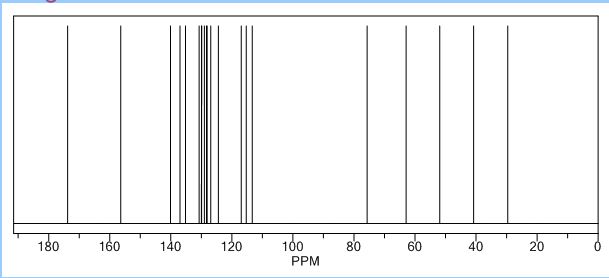
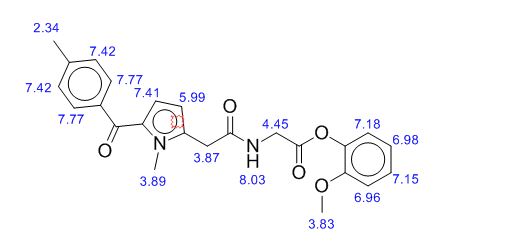
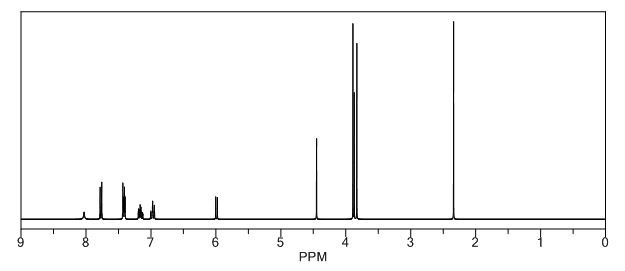
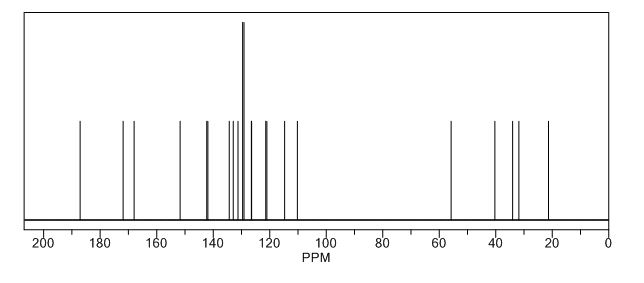
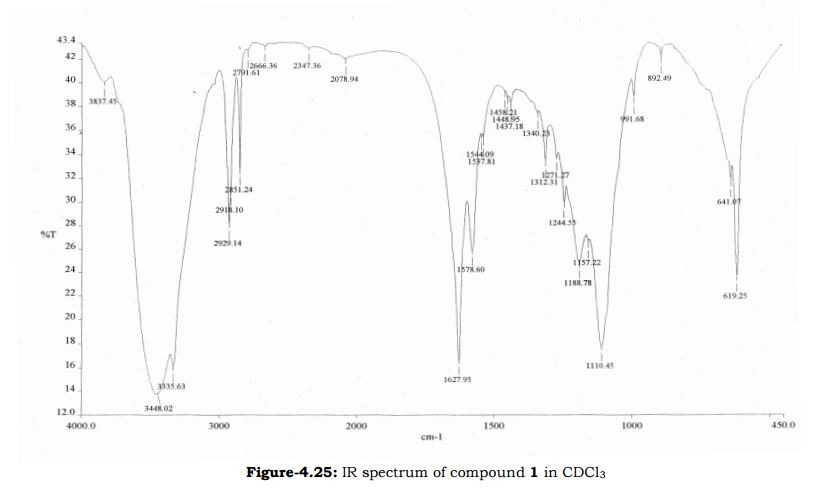
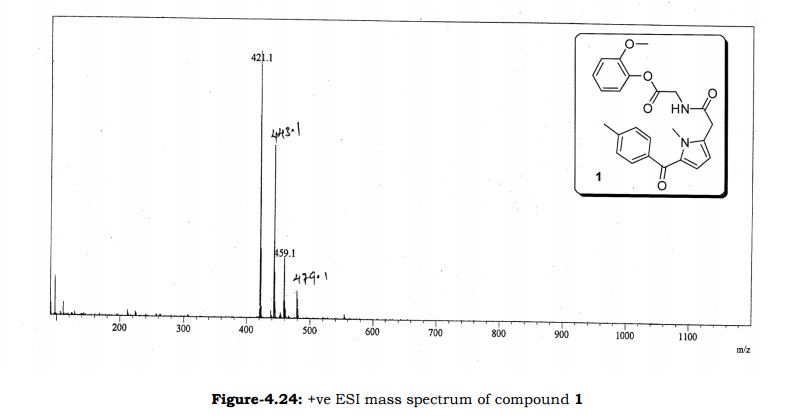
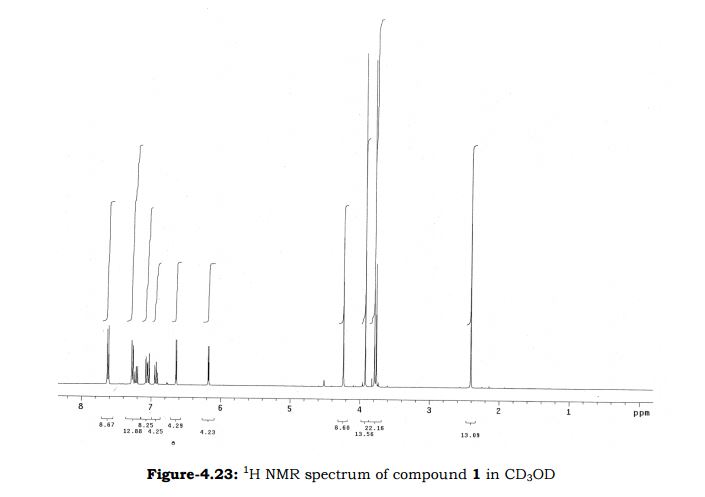
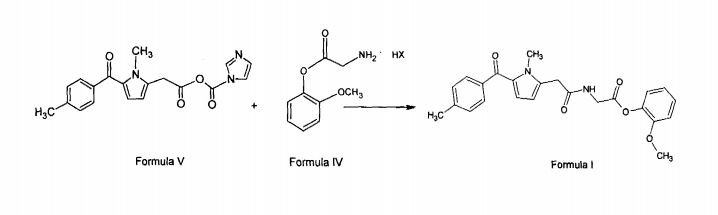
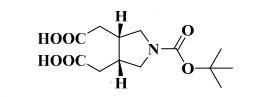
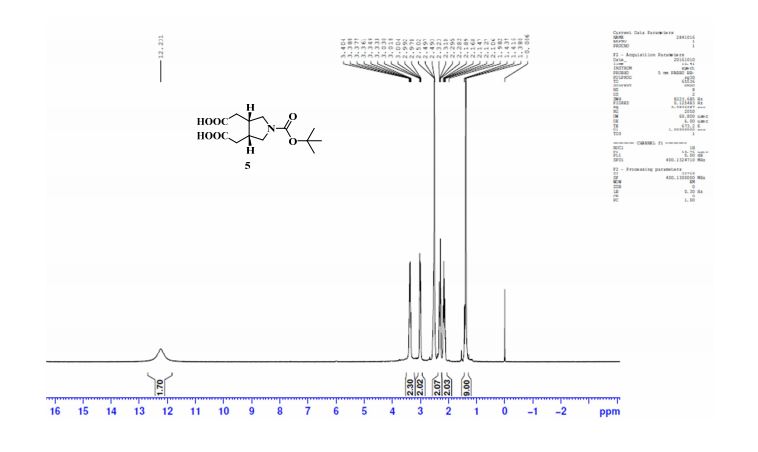
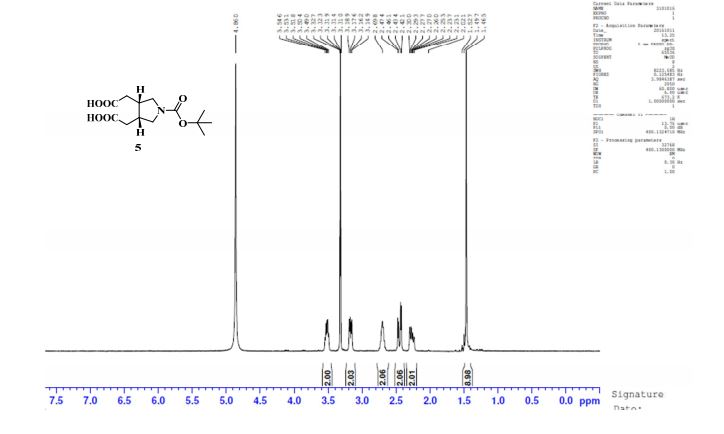
Study on the Isolation and Chemical Investigation of Potential Impurities in Dexrazoxane Using 2D-NMR and LC-PDA-MSImage may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.Image may be NSFW. Clik here to view.
§ Centre for Chemical Science & Technology, Institute of Science & Technology, Jawaharlal Nehru Technological University, Hyderabad 500085, Telangana, India
†Gland Pharma Ltd, Research and Development, D.P.Pally, Hyderabad 500043, Telangana, India
A chemical investigation of process related impurities associated with the synthesis of dexrazoxane was performed. The degradation product of dexrazoxane is known in the literature; however, the information related to process impurities is not available. For the first time, two process related impurities were isolated, characterized, and confirmed by their individual chemical synthesis. The present study describes the isolation methods of the impurities and their structural elucidation using IR, 1H, 13C, 2D NMR, and mass spectrometry. The identification of these impurities would be highly valuable for the quality control during the production of the dexrazoxane drug substance
The U.S. Food and Drug Administration (FDA)(5) and the European Medicine Agency (EMA)(6) require analytical characterization not only for the active pharmaceutical ingredient (API), but also for its key starting materials and advanced intermediates. The determination of a drug substance impurity profile, including especially known pharmacopeial impurities, as well as other unknown impurities, could have a significant impact on the quality and the safety of the drug products. The health implications of the impurities could be significant because of their potential mutagenic or carcinogenic effects. Therefore, the International Conference on Harmonization (ICH) has set a high standard for the purity of drug substances.(7) If the dose is less than 2 g/day, then impurities over 0.10% are expected to be identified, qualified, and controlled. If the dose exceeds 2 g/day, then the qualification threshold is lowered to 0.05%. It is therefore essential to monitor and control the impurities in both the drug substance (API) and the drug products.
5 Guidance for Industry on Abbreviated New Drug Applications: Impurities in Drug Substances; Availability;Fed. Regist.2009, 74, 34359– 34360.
6.The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Impurities in New Drug Substances Q3A (R2); IGH: Geneva, Switzerland, 2006.
7.International Conference on Harmonization; revised guidance on Q3A impurities in new drug Substances; Availability; Fed. Regist.2003, 68, 6924– 6925.
The transposition of Sandmeyer chlorination from a batch to a safe continuous-flow process was investigated. Our initial approach was to develop a cascade method using flow chemistry which involved the generation of a diazonium salt and its quenching with copper chloride. To achieve this safe continuous process diazotation, a chemometric approach (Simplex method) was used and extrapolated to establish a fully continuous-flow method. The reaction scope was also examined via the synthesis of several (het)aryl chlorides. Validation and scale-up of the process were also performed. A higher productivity was obtained with increased safety.
Efficient Transposition of the Sandmeyer Reaction from Batch to Continuous Process
The reaction was carried out as described in general procedure B using 2-Amino-5-chlorobenzoxazole (221 mg, 1.31 mmol). After purification with silica flash chromatography (EP 100%), the product was isolated as a yellow oil (62 mg, 25%).
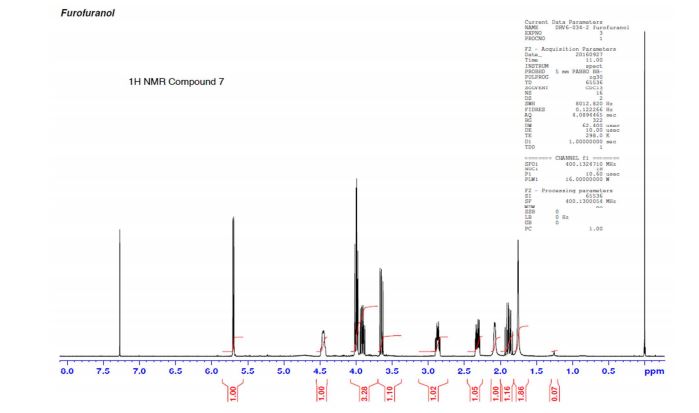
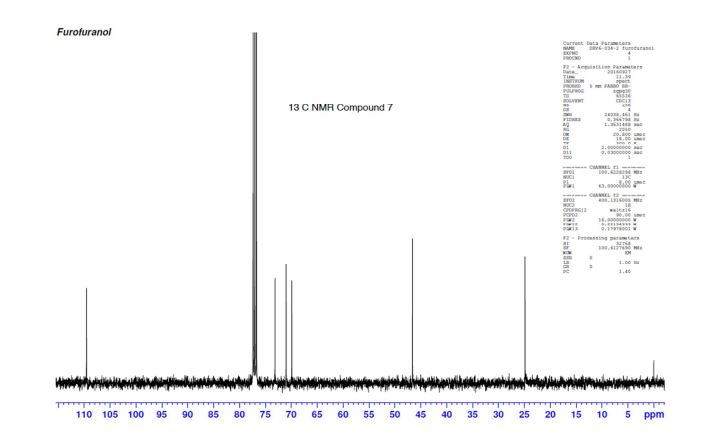
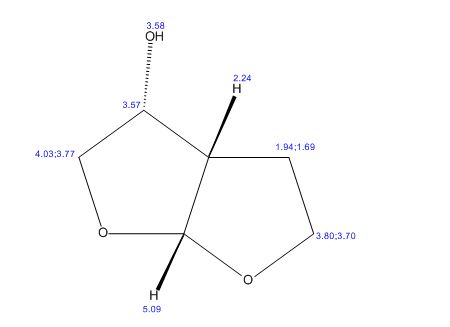
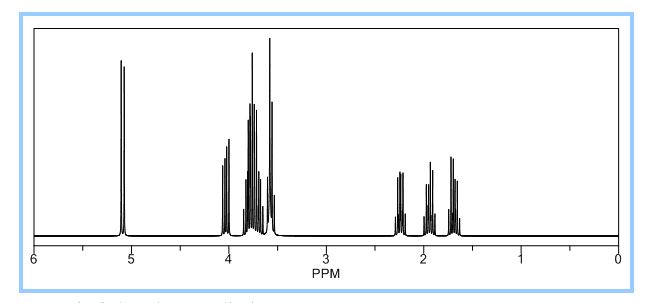
A practical synthesis of (3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-ol—a key intermediate in the synthesis of darunavir—from monopotassium isocitrate is described. The isocitric acid salt, obtained from a high-yielding fermentation fed by sunflower oil, was converted in several steps to a tertiary amide. This amide, along with the compound’s ester functionalities, was reduced with lithium aluminum hydride to give, on acidic workup, a transient aminal-triol. This was converted in situ to the title compound, the bicyclic acetal furofuranol side chain of darunavir, a protease inhibitor used in treatment of HIV/AIDS. Key to the success of this process was identifying an optimal amide that allowed for complete reaction and successful product isolation. N-Methyl aniline amide was identified as the most suitable substrate for the reduction and the subsequent cyclization to the desired product. Thus, the side chain is produced in 55% overall yield from monopotassium isocitrate.
Practical Synthesis of the Bicyclic Darunavir Side Chain: (3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-ol from Monopotassium Isocitrate
In particular, the following synthetic scheme (1) illustrates the present commercial method of synthesizing compound (I) . This synthesis is disclosed in detail in A.K. Ghosh et al . , Tetra- hedron Letters, 36 (4) , pp. 505-508 (1995), incorporated herein by reference. Also see, A.K. Ghosh et al., J. Med. Chem . , 39, pp. 3278-3290 (1996) for the synthesis of compound (I) and a related compound of structural formula (II) (i.e., (3S, 3aR, 7aS) -3- hydroxyhexahydrofuro [2, 3-b] pyran) .
2. NaBH4, EtOH (65-75%) -15°C, 1-3 h Compound (I) (bis-THF) Another aspect of the present invention is to provide a method of preparing a compound having a structure
then utilizing the benzyl-protected 5-hydroxymethyl- 5H-furan-2-one in the synthesis of compound (I) .
Another aspect of the present invention is to provide a method of preparing compounds related to bis-THF by using a starting material having a following structure:
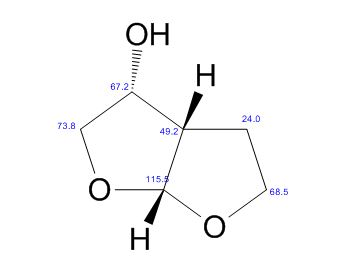
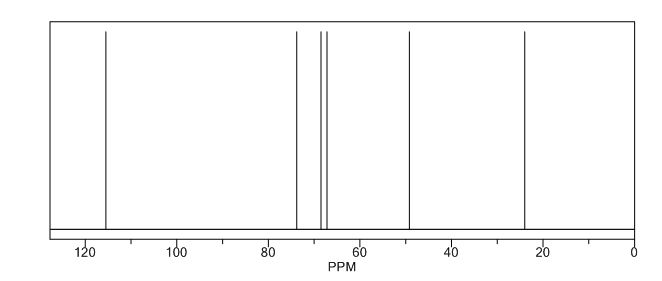
(3R, 3aS, 6aR) -3-Hydroxyhexahydrofuxo [2, 3- b] furan (I) : To a solution containing 250 mg (1.95 mmol) (3aS, 6aR) -3-oxyhexahydrofuro [2, 3-b] furan (6) in EtOH (25 mL) was added ’89 mg (2.35 mmol) NaBH4 at -18 °C. The reaction mixture was stirred at -18 °C for 2.5 hours, then the reaction was quenched with saturated NH4C1 solution (5 mL) and warmed to room temperature. The resulting mixture was concentrated under reduced pressure, and then 10 mL water was added. The aqueous layer was extracted with ethyl acetate (3 x 50 mL) and a solution of 70% CHC13, 20% MeOH, and 10% water (3 x 50 mL) . The combined organic extracts were dried over Na2S04. Column chromatography (silica gel 80 g, MeOH in CHC13 7%) gave compound (I) (178 mg. 70%) as a colorless solid, Rf=0.3, [α]25D -12.4°, c 1.3, MeOH. IR (neat) 2951, 1641, 1211 cm“1; XH-NMR (400 MHz CDC13) δ: 1.85 (mc, IH) , 1.94 (bs, IH) , 2.27 (mc, IH) , 2.84 (mc, IH) , 3.63 (dd, IH, J=7.1 Hz, J=9.2 Hz), 3.89 (mc, IH) , 3.97 (mc, IH) , 4.43 (dd, IH, J=6.8 Hz, J=14.5 ” Hz), 5.68 (d, IH, J=5.2 Hz). 13C-NMR (125.8 MHz, CDC13, Dept) δ‘: 25.27 (-) , 46.97 (+) , 70.31 (-) , . 71.26 (-), 73.50 (+) , 109.93. (+) . C6H10O3; Exact Mass: 130.06; Mol. Wt . : 130.14; C, 55.37, H, 7.74, 0, 36.88.
Experimentals :
l-(Benzyloxy)-but-3-en-2-ol (±)-(8): To a solution of vinylmagnesium bromide (1 M in THF, 40 mL, 40 mmol) in THF (10 mL) at 0°C was added benz- yloxyacetaldehyde (7) (5 g, 33.3 mmol) dropwise. The mixture was stirred for 10 min at 0°C, and the reaction then was quenched with 20 L of saturated NaHC03 solution. The layers were separated, the aqueous layer was extracted with ethyl acetate (3 x 20 mL) , and the combined organic extracts were dried over sodium sulfate. Evaporation of solvent under reduced pressure, followed by column chromatography on silica gel (20% EtOAc in hexanes as the eluent) yielded alcohol (±)-8 (5.22 g, 88%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes); 1H-NMR (400 MHz, CDC13) δ: a 2.79 (bs, IH) , 3.39 (dd, IH, J=1.7, 7.85 Hz), 3.55 (dd, IH, J=3.35, 6.3 Hz), 4.35 (m, IH) , 4.58 (s, IH) , 5.21 (dt, IH, J=7.75, 1.4 Hz), 5.38 (dt, IH, J=14.18, 1.4 Hz), 5.84 (m, IH) , 7.30-7.38 (m, 5H) ; 13C-NMR (100.6 MHz, CDC13) δ: 71.52, 73.37, 74.02, 116.49, 127.85, 128.49, 136.58, 137.81. (S)-l-(Benzyloxy) -but-3-en-2-ol (9) and (R) -1- (benzyloxy) -but-3-en-2-oyl acetate (10):
A: To a solution of alcohol (±)-(8) (5.21 g, 29.3 mmol) in acetic anhydride (14 mL, 147 mmol) and tert-butyl methyl ether (70 mL, 586 mmol) was added immobilized lipase PS-30 (5.3 g ) on Celite 521 (Aldrich) . The mixture was stirred at room temperature for 20 h, and then filtered through Celite. Removal of solvent under reduced pressure followed, by column chromatography on silica gel (10 and 15% EtOAc in hexanes as the eluents) yielded acetate (10) (3.81 g, 54%) Rf=0.57 (30% EtOAc in hexanes) as a clear oil, [of]25D -2° (c 1, CHC13) ; NMR (500 MHz, CDC13) δ: 2.10 (s, 3H) , 3.55-3.59 (m, 2H) , 4.56 (q, 2H, J=12.2, 14.0 Hz), 5.24 (d, IH, J-10.6 Hz), 5.32 (d, IH, J=17.3 Hz), 5.50 (m, IH) , 5.84 (m, IH) , 7.25-7.36 (m, 5H) ; 13C-NMR (125.8 MHz, CDC13) δ: 21.62, 71.67, 73.57, 73.59, 118.39, 128.14, 128.84, 133.77, 138.32, 170.63; alcohol 9 (2.34 g, 45%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [α]25D– 8.3° (c 1.06, MeOH) .
B: To a solution of alcohol (±)-(8) (3.92 g, 22.0 mmol) in vinyl acetate (46 mL, 499 mmol) and ethylene glycol dimethyl ether (46 mL, 440 mmol) was added immobilized lipase PS-30 (4 g ) on Celite-545 (Aldrich) . The mixture was stirred at room temperature for 28 h, and then filtered through celite. Removal of solvent under reduced pressure, followed by column chromatography on silica gel (10 and 15% EtOAc in hexanes as the eluents) yielded acetate
(10) (2.20 g, 45%) Rf=0.57 (30% EtOAc in hexanes) as a clear oil, [ ]25D -2.7° (c 1.35, MeOH); alcohol (9) (2.00 g, 51%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [α]25D -11.4° (c 1.6, MeOH).
C: To a solution of alcohol (+)-(8) (30 mg, 0.168 mmol) in isopropenyl acetate (375 μL, 3.36 mmol) and ethylene glycol dimethyl ether (375 μL, 3.61mmol) was added immobilized lipase PS-30 (35 mg) on Celite-545 (Aldrich) . The mixture was stirred at room temperature for 23 h, and then filtered through celite. Removal of solvent under reduced pressure, followed by column chromatography on silica gel (10) and 15% EtOAc in hexanes as the eluents) yielded acetate 10 (20.3 mg, 54%) as an oil, Rf=0.57 (30% EtOAc in hexanes), [α]25D -1.4° (c 1.02, MeOH); alcohol (9) (13 mg, 43%) as a yellow oil, Rf=0.40
(30% EtOAc in hexanes), [ ]25D -13.5° (c 1.3, MeOH). (R) -1- (Benzyloxy) -but-3-en-2-ol (11): To a solution of acetate (10) (3.7 g, 16.9 mmol) in methanol (20 mL) was added K2C03 (7 g, 50.6 mmol). The mixture was stirred at room temperature for 35 min. Methanol then was removed under reduced pressure. The resulting solid residue was dissolved in ethyl acetate, washed with saturated NH4C1 solution and brine, and dried over sodium sulfate. Removal of ethyl acetate under reduced pressure yielded the crude alcohol (11) (3 g, 100%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [α]25D 8.3° (c 1.06, MeOH) .
(S)-l- (Benzyloxy) -but-3-en-2-ol (9) from (11): To a solution of crude alcohol (5) (2 g, 11.2 mmol), triphenylphosphine (5.88 g , 22.4 mmol), and 4-nitrobenzoic acid (2.81 g, 16.8 mmol) in benzene (35 mL) was added at room temperature diisopropyl azodicarboxylate (4.35 mL, 22.4 mmol) dropwise. The mixture was stirred for 40 min, followed by the re- moval of solvent under reduced pressure. All of the crude ester then was dissolved in a mixture of MeOH:Et3N:H20 (20ml) in the ratio of 4:3:1 and reacted with LiOH (1.64 g, 39.3 mmol) at room temperature. The mixture was stirred for 2 h, followed by the removal of solvent. Column chromatography on silica gel (15% EtOAc in hexanes as the eluent) yielded alcohol (3) (1.64 g, 82%) as a yellow oil, Rf=0.40 (30% EtOAc in hexanes), [o;]25D -7.3° (c 0.82, MeOH) . (S) -1- (Benzyloxy) -but-3-en-2-yl acrylate
(12): To a solution of alcohol (3) (1 g, 5.61 mmol) in CH2C12 (20 L) was added acryloyl chloride (685 μL, 8.41 mmol) dropwise, followed by the addition of Et3N (1.56 mL, 11.2 mmol). The resulting mixture was stirred for 10 min, and the solvent then was removed under reduced pressure. Filtration of the concentrated crude acrylate through a pad of silica gel using 15% EtOAc in hexanes, followed by the removal of solvent, yielded acrylate (12) (1.19 g, 92%) as a colorless oil, Rf=0.57 (30% EtOAc in hexanes), [α]25D -5.7° (c 1.09, CHC13) ; 1H-NMR (500 MHz, CDC13) δ: 3.59-3.65 (m, 2H) , 4.56 (q, 2H, J=12.2, 14.65 Hz), 5.25 (d, IH, J=10.6 Hz), 5.33 (d, IH, J=16.8 Hz), 5.57 (m, IH) , 5.84-5.91 (m, 2H) , 6.17 (dd, IH, J=6.9, 10.4 Hz), 6.44 (dd, IH, J=1.3, 16.2 Hz), 7.27-7.36 (m, 5H) ; 13C-NMR (125.8 MHz, CDC13) δ: 71 . 62 , 73 . 58 , 73 . 77 , 118 . 49 , 128 . 05 , 128 . 85 , 131 . 52 , 133 . 62 , 138 . 31 , 165 . 79 .
(5S) -5- (Benzyloxymethyl) -5H-furan-2-one (13): To a solution of acrylate (12) (1.87 g, 8.05 mmol) in CH2C12 (700 mL) was added second generation Grubbs’ catalyst (4 mol %, 170 mg, 0.322 mmol). The reaction mixture was refluxed for 5 hours, and the solvent then was removed under reduced pressure. Column chromatography on silica gel (30% EtOAc in hexanes as the eluent) yielded the furanone (13)
(4S ,5S) -5- (Benzyloxymethyl) -4- [1 , 3] di- oxolan-2-yldihydrofuran-2-one (14) : A solution of furanone (13) (1.2 g, 5.88 mmols) and benzophenone
(108 mg, 0.588 mmols) in [1, 3] -dioxolane (108 mg) was degassed for 40 min in a stream of argon. The mixture then was irradiated using one 450 watt ACE glass medium pressure mercury lamp, from a distance of 15 cm, for 9 hours. Progress of this reaction was observed via 1H-NMR. As the reaction mixture was degassed, and throughout all of the irradiation time, the reaction flask was held in a water cooled cooling mantel. The temperature of the cooling water was constantly maintained near 0°C. Upon completion of the reaction, solvent was removed under reduced pressure, followed by column chromatography on silica gel (35% EtOAc in hexanes as the eluent), yielding the title compound (1.34 g, 82%) as a clear oil, Rf=0.14 (30% EtOAc in hexanes), [α]25D 16.5° (c 1.2, CHC13) ; 1H-NMR (500 MHz, CDC13) δ: 2.50 (dd, IH, J=3.9, 12.9 Hz), 2.70-2.79 (m, 2H) , 3.58 (dd, IH, J=3.5, 7.2 Hz), 3.75 (dd, IH, J=2.8, 7.9 Hz), 3.87-3.92 (m, 2H) , 3.97-4.00 ( , 2H) , 4.51 (d, IH, J=11.9 Hz), 4.57-4.61 (m, 2H) , 4.88 (d, IH, J=3.6 Hz), 7.26-7.36 (m, 5H) ; 13C-NMR (125.8 MHz, CDC13) δ: 30.39, 40.53, 65.77, 71.74, 73.99, 79.52, 104.14, 128.00, 128.89, 138.07, 176.79.
(4S,5S) -4-[l,3]Dioxolan-2-yl-5-hydroxy- methyldihydrofuran-2-one (15) : To a solution of dihydrofuranone (14) (0.5 g, 1.79 mmol) in MeOH (30 mL) was added Pd/C (25 mg) . The mixture was stirred at room temperature under an H2 balloon for 24 hours, and then filtered over Celite. Removal of solvent under reduced pressure, followed by column chromatography on silica gel (35% EtOAc in hexanes as the eluent) yielded the compound (15) (301 mg, 89%) as a white solid, Rf=0.28 (50% EtOAc in hexanes), [ ]25D 22° (c 1.32, CHC13) ; XH-NMR (500 MHz, CDC13) δ: 2.54 (dd, IH, J=6.0, 11.4 Hz), 2.68-2.81 (m, 2H) , 3.66
(dd, IH, J=3.9-8.5 Hz), 3.88-3.95 (m, 3H) , 3.97-4.02 (m, 2H) , 4.53 (m, IH) , 4.91 (d, IH, J=3.9 Hz); 13C- NMR (125.8 MHz, CDC13) δ: 30.68, 40.12, 64.36, 65.77, 81.07, 103.94, 176.83. (3S , 3aS , 6aR) -3-Hydroxyhexahydrofuro [2 , 3- b] furan (5) : To a solution of lithium aluminum hydride (76 mg, 1.98 mmols) in THF (10 ml) at 0°C was added dihydrofuranone 15 (275 mg , 1.46 mmol) in THF (30 mL ) dropwise. Upon completion of the reduction after 4 hours, the reaction was quenched with a saturated aqueous sodium sulfate solution at 0°C. The solvent then was decanted and the remaining residue was washed with THF (3x) , EtOAc (3x) , and CHC13 (3x) . The organic extracts were combined and the solvent was removed under reduced pressure, yielding a crude (2S, 3S) -3- [1, 3] dioxolan-2- ylpentane-1, 2, 5-triol, which was immediately used in the next reaction.
The crude triol was dissolved in a mixture of THF:H20 (8ml) in the ratio of a 5:1. This solu- tion then was acidified at room temperature to pH 2- 3 with 1 N hydrochloric acid, and was stirred for 40 hours. Removal of solvent with the aid of benzene under reduced pressure, followed by column chromatography purification on silica gel (5% MeOH in CHCI3 as the eluent) yielded the compound (5) (145 mg,
77%) as a white solid, Rf=0.40 (15% MeOH in CHC13) , [α]25D -25.1° (c 1.05, CHC13) ; XH-NMR (500 MHz, CDCI3) δ: 1.67 ( , IH) , 2.13 (m, IH) , 2.31 (bs, IH) , 2.79 (m, IH) , 3.80-3.88 (m, 3H) , 3.95 (dd, IH, J=3.2, 7.1 Hz), 4.20 (d, IH, J=3.1 Hz), 5.86 (d, IH, J=4.9 Hz). Preparation of bis-THF derivative (I) (by Mitsunobu inversion of compound (5) ) : To a stirred solution of alcohol (5) (400 mg, 3.07 mmol), tri- phenylphosphine (1.6 g, 61.4 mmol), and p-nitroben- zoic acid (770 mg, 4.61 mmol) in dry benzene (30 mL) at 23 °C was added diisoproylazodicarboxylate (DIAD, 1.2 L, 6.14 mmol) dropwise. After 1.5 hours, the mixture was concentrated in vacuo, and the crude ester was dissolved in a (4:3:1) mixture of MeOH:Et3N:H20 (24 mL) , then treated with LiOH (450 mg, 10.7 mmol) . The solution was stirred at room temperature for 2 h. The mixture then was concentrated under reduced pressure and the residue was chromatographed over silica gel to provide the bis-
In particular, the following synthetic scheme (1) illustrates the present commercial method of synthesizing compound (I). This synthesis is disclosed in detail in A.K. Ghosh et al., Tetra. hedron Letters, 36 (4) , pp. 505-508 (1995), incorporated herein by reference. Also see, A.K. Ghosh et al., J. Med. Chem . , 39, pp. 3278-3290 (1996) for the synthesis of compound (I) and a related compound of structural formula (II) (i.e., (3S, 3aR, 7aS) -3-hydroxyhexahydrofuro [2, 3-b] pyran).
Image may be NSFW. Clik here to view.
Image may be NSFW. Clik here to view.
The present method of synthesizing bis-THF is summarized as follows:
Image may be NSFW. Clik here to view.
Image may be NSFW. Clik here to view.
The synthesis of bis-THF (compound (I) ) is summarized below:
GMP’s for Early Stage Development of New Drug substances and products
The question of how Good Manufacturing Practice (GMP) guidelines should be applied during early stages of development continues to be discussed across the industry and is now the subject of a new initiative by the International Consortium on Innovation and Quality in Pharmaceutical Development (IQ Consortium)—an association of pharmaceutical and biotechnology companies aiming to advance innovation and quality in the development of pharmaceuticals. They have assembled a multidisciplinary team (GMPs in Early Development Working Group) to explore and define common industry approaches and to come up with suggestions for a harmonized approach. Their initial thoughts and conclusions are summarized in Pharm. Technol. 2012, 36 (6), 54–58.
Image may be NSFW. Clik here to view.
From an industry perspective, it is common to consider the “early” phase of development as covering phases 1 and 2a clinical studies. During this phase, there is a high rate of product attrition and a high probability for intentionally introducing change into synthetic processes, dosage forms, analytical methods, and specifications. The quality system implemented during this early phase should take into account that these changes and adjustments are intrinsic to the work being performed prior to the determination of the final process and validation of the analytical methods during later stages of development.
Image may be NSFW. Clik here to view.
FDA guidance is already available on GMP requirements for phase 1 materials. (See Org. Process. Res. Dev. 2008, 12, 817.) Because many aspects of phase 2a clinical studies are similar in their scope and expectations, the working group feels there is an opportunity to extend this guidance across all early phase studies. Because products and processes are less well understood in the early phases of development, activities should focus on accumulating the appropriate knowledge to adequately ensure patient safety. Focusing on this area should ensure that beneficial therapies reach the clinic in an optimum time scale with minimal safety concerns.
Image may be NSFW. Clik here to view.
A follow-up article ( Pharm. Technol. 2012, 36 (7), 76−84) describes the working group’s approach to the subject of Analytical Method Validation. Their assessment has uncovered the need to differentiate the terms “validation” and “qualification”. Method qualification is based on the type, intended purpose, and scientific understanding of the type of method in use. Although not used for GMP release of clinical materials, qualified methods are reliable experimental methods that may be used for characterization work such as reference standards and the scientific prediction of shelf life. For example, in early development it would be sufficient for methods used for in-process testing to be qualified, whereas those methods used for release testing and for stability determination would be more fully validated.
In early development, a major purpose of analytical methods is to determine the potency of APIs and drug products to ensure that the correct dose is delivered in the clinic. Methods should also indicate stability, identify impurities and degradants, and allow characterization of key attributes. In the later stages, when processes are locked and need to be transferred to worldwide manufacturing facilities, methods need to be cost-effective, operationally viable, and suitably robust such that the methods will perform consistently. irrespective of where they are executed.
The authors advocate that the same amount of rigorous and extensive method-validation experiments, as described in ICH Q2, “Analytical Validation”, is not needed for methods used to support early stage drug development. For example, parameters involving interlaboratory studies (i.e., intermediate precision, reproducibility, and robustness) are not typically performed during early phase development, being replaced by appropriate method-transfer assessments and verified by system suitability requirements. Because of changes in synthetic routes and formulations, the impurities and degradation products formed may change during development.
Accordingly, related substances are often determined using area percentage by assuming that the relative response factors are similar to that of the API. As a result, extensive studies to demonstrate mass balance are typically not conducted during early development.
Detailed recommendations are provided for each aspect of method validation (specificity, accuracy, precision, limit of detection, limit of quantitation, linearity, range, robustness) according to the nature of the test (identification, assay, impurity, physical tests) for both early- and late phase development. These recommendations are also neatly summarized in a matrix form.
Due to the high attrition rate in early development, the focus should be on consistent specifications that ensure patient safety, supported by preclinical and early clinical safety studies. On the basis of the cumulative industry experience of the IQ working group members, the authors of this paper propose standardized early phase specification tests and acceptance criteria for both drug substance and drug product. In addition to release and stability tests, consideration is given to internal tests and acceptance criteria that are not normally part of formal specifications, but which may be performed to collect information for product and process understanding or to provide greater control.
Image may be NSFW. Clik here to view.
The drug substance used in preclinical animal studies (tox batch) is fundamental in defining the specifications for an early phase clinical drug substance (DS). Here, internal targets rather than formal specifications are routinely used while gathering knowledge about impurities and processing capabilities. At this stage the emphasis should be on ensuring the correct DS is administered, determining the correct potency value, and quantitating impurities for toxicology purposes. For DS intended for clinical studies, additional testing and controls may be required; the testing may be similar to that for the tox batch, but now with established acceptance criteria. For these stages the authors propose a standardized set of DS specifications, as follows.
Description
range of colour
identification
conforms to a reference spectrum
counterion
report results
assay
97–103% on a dry basis
impurities
NMT 3.0% total, NMT 1.0% each
unidentified
NMT 0.3%
unqualified
NMT 0.15%
mutagenic
follow EMA guidelines (pending ICH M7 guidance)
inorganic
follow EMA guidelines (pending ICH Q3D guidance)
residual solvents
use ICH Q3C limits or other justified limits for solvents used in final synthetic step
water content
report results
solid form
report results
particle size
report results
residue on ignition
NMT 1.0%
These may be altered in line with any specific knowledge of the compound in question. For example, if the DS is a hydrate or is known to be hygroscopic or sensitive to water, a specified water content may be appropriate. Of particular note is the use of impurity thresholds which are 3 times higher than those defined in ICH Q3 guidelines. Q3 was never intended to apply to clinical drugs, and higher thresholds can be justified by the limited exposure that patients experience during these early stages. Mutagenic impurities are the exception here, since in this area the existing official guidance does cover clinical drugs.
The fourth article in the series(Acken, B.; Alasandro, M.; Colgan, S.; Curry, P.; Diana, F.; Li, Q. C.; Li, Z. J.; Mazzeo, T.; Rignall, A.; Tan, Z. J.; Timpano, R.Early Development GMPs for Stability (Part IV)Pharm. Technol.2012, 36 ( 9) 64– 70) considers appropriate approaches to stability testing during early clinical phases. Appropriate stability data at suitable storage conditions are required to support filing the clinical trial application (CTA/IND/IMPD) and use of the clinical material through the end of the clinical study. Several factors from business, regulatory, and scientific perspectives need to be taken into account when designing early stability studies, such as the risk tolerance of the sponsoring organization, the inherent stability of the drug substance and prior product, process and stability knowledge, the regulatory environment in the countries where the clinical trial will be conducted, and the projected future use of the product.
Often non-GMP DS batches are manufactured first and placed on stability to support a variety of product development activities.In many cases these batches will be representative of subsequent GMP batches from a stability perspective and can be used to establish an initial retest period for the DS and support a clinical submission. In early development, it is common for the manufacturing process to be improved; therefore, as the DS process evolves, an evaluation is needed to determine whether the initial batch placed on stability is still representative of the improved process. The authors advocate a science- and risk-based approach for deciding whether stability studies on new process batches are warranted.
The first step is to determine which DS attributes have an effect on stability. This step can be completed through paper-based risk assessments, prior knowledge, or through a head-to-head short-term stability challenge. If the revised process impacts one or more of these stability-related quality attributes, the new batch should be placed on stability—otherwise not. Typical changes encountered at this stage include changes in synthetic pathway, batch scale, manufacturing equipment or site, reagents, source materials, solvents used, and crystallization steps.
Image may be NSFW. Clik here to view.
In most cases, these changes will not result in changes in DS stability. Changes to the impurity profile are unlikely to affect stability, since most organically related impurities will be inert. On the other hand, catalytic metals, acidic or basic inorganic impurities, or significant amounts of residual water or solvents may affect stability; thus, changes to these attributes would typically require the new batch to be placed in the stability program. Similarly, any changes to polymorphic form, particle size, or counterion would warrant extra testing. Packaging changes of the bulk material to a less protective package may require stability data to support the change.
Three approaches to stability data collection are commonly used. One is that an early, representative DS batch is placed under real-time and accelerated conditions (e.g., 25 °C/60% RH and 40 °C/75% RH), and stability results for a few time points (e.g., 1–6 months) are generated to support an initial retest period (e.g., 12 months or more). A second approach is to use high stress conditions such as a high temperature and high humidity with a short time. A third approach is the use of stress studies at several conditions coupled with modelling. The retest period derived from these types of accelerated or stress studies can be later verified by placing the first clinical batch into real-time stability studies under ICH accelerated and long-term conditions. Future extensions of the retest/use period can be based on real-time data.
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
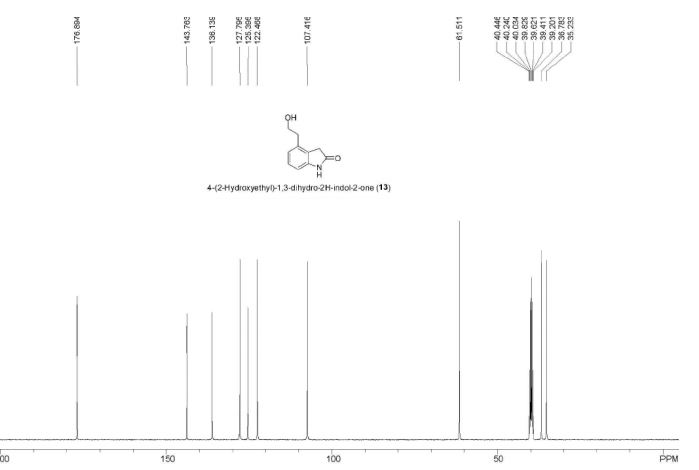
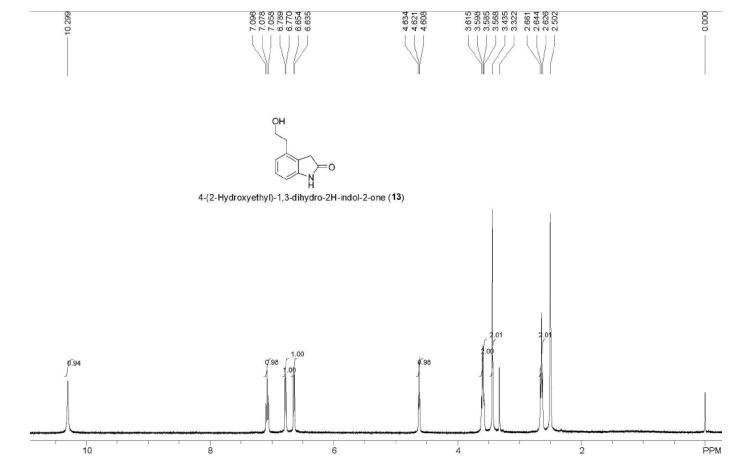
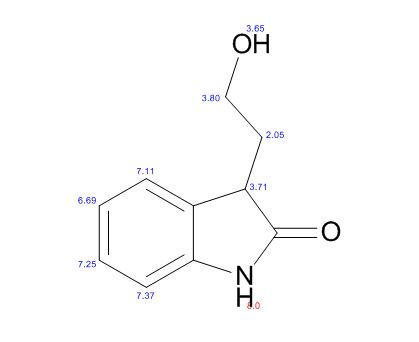
ESI-MS (m/z) 178 [M + H]+. Anal. Calcd for C10H11NO2: C, 67.78; H, 6.26; N, 7.90. Found: C, 67.73; H, 6.20; N, 7.82.
Image may be NSFW. Clik here to view.
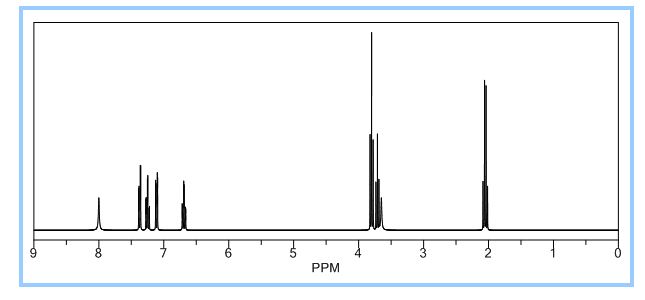
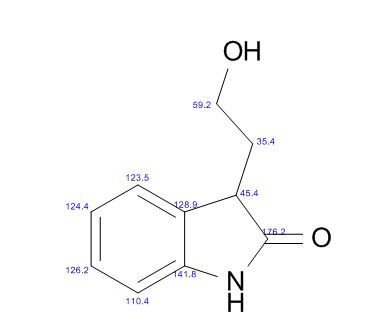
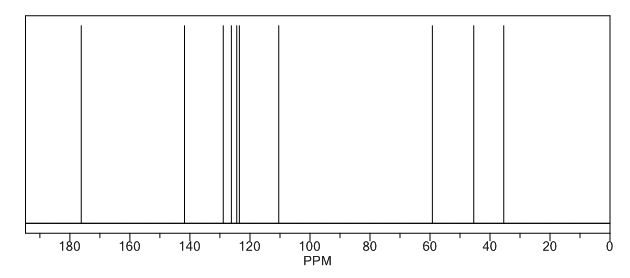
A new and efficient manufacturing technology is disclosed in the present work for the preparation of 4-(2-hydroxyethyl)-1,3-dihydro-2H-indol-2-one, which is a key intermediate for ropinirole hydrochloride. The whole process gives the target molecule in 71% overall yield with 99% purity. In the final step, a novel nitro reduction/ring-closing/debenzylation takes place in one pot. All the intermediates can be used directly for the next step without purification in this process.
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
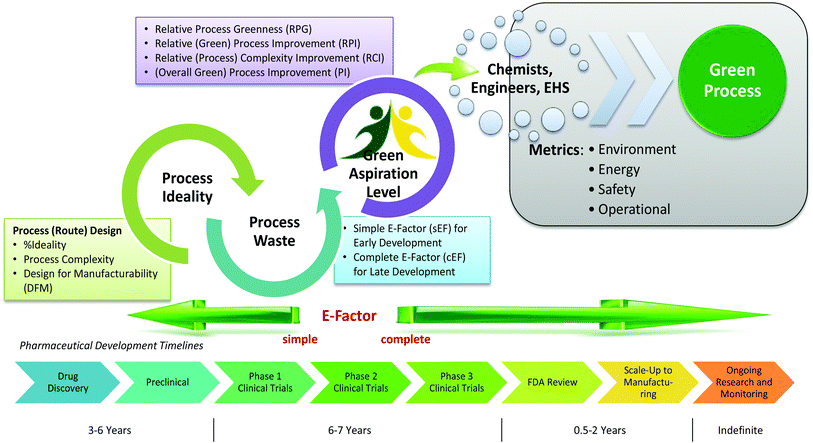
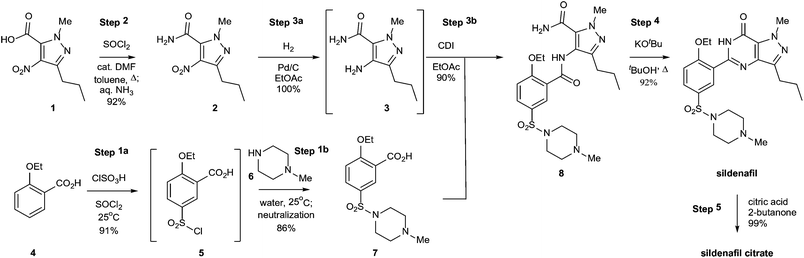
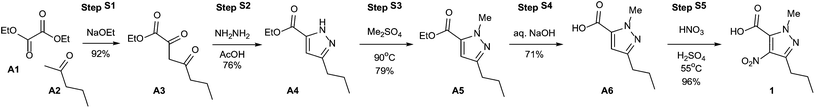
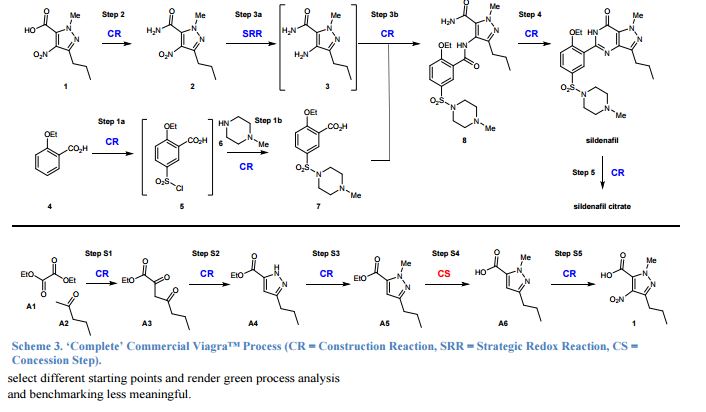
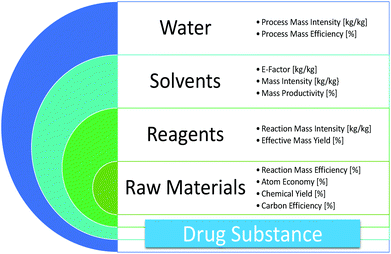
“Green chemistry” refers to the promotion of safe, sustainable, and waste-minimizing chemical processes. The proliferation of green chemistry metrics without any clear consensus on industry standards is a significant barrier to the adoption of green chemistry within the pharmaceutical industry. We propose the Green Aspiration Level™ (GAL) concept as a novel process performance metric that quantifies the environmental impact of producing a specific pharmaceutical agent while taking into account the complexity of the ideal synthetic process for producing the target molecule. Application of the GAL metric will make possible for the first time an assessment of relative greenness of a process, in terms of waste, versus industry standards for the production process of any pharmaceutical. Our recommendations also include a simple methodology for defining process starting points, which is an important aspect of standardizing measurement to ensure that Relative Process Greenness (RPG) comparisons are meaningful. We demonstrate our methodology using Pfizer’s Viagra™ process as an example, and outline aspiration level opportunities for industry and government to dismantle green chemistry barriers.
Image may be NSFW. Clik here to view.
Overcoming barriers to green chemistry in the pharmaceutical industry – the Green Aspiration Level™ concept
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
///////////green chemistry, pharmaceutical industry
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
CrystEngComm, 2017, 19,335-345 DOI: 10.1039/C6CE01645F, Paper
Ruimiao Chang, Qiang Fu, Yong Li, Mingchan Wang, Wei Du, Chun Chang, Aiguo Zeng
In this paper, four amorphous samples of loratadine were prepared by quench-cooling the melted drug at different temperatures.
In this paper, four amorphous samples of loratadine were prepared by quench-cooling the melted drug at different temperatures. With these samples, the crystallization tendencies were tested by powder X-ray diffraction (PXRD), and non-isothermal cold crystallization kinetics was investigated by using differential scanning calorimetry (DSC) and the molecular dynamics both in super-cooled liquid and in glassy states was analyzed by using broadband dielectric spectroscopy (BDS) at a temperature range from 213 to 393 K. From the PXRD results, it was established that the four amorphous loratadine samples were apt to crystallize at a temperature below the glass transition temperature. From the DSC results, it was found that the non-isothermal crystallization mechanism of these four loratadine forms was similar. However, the fast crystallization tendency (low physical stability) was also observed for the amorphous loratadine which was obtained at a low quench-cooling temperature. The tendency was analyzed based on the BDS results which demonstrated that rapid molecular mobility could generate a low physical stability and was closely related to Johari–Goldstein relaxation. These results suggested that loratadine had a weak frustration against crystallization and its physical stability was affected by the quench-cooling temperature. This study laid a foundation for choosing the right technique to prepare the amorphous form of loratadine and improving its physical stability.
Crystallization and relaxation dynamics of amorphous loratadine under different quench-cooling temperatures
MEDOSAN RICERCA S.R.L. [IT/IT]; Via Cancelliera, 12 I-00040 Cecchina RM (IT) (For All Designated States Except US). SIGMA-TAU INDUSTRIE FARMACEUTICHE RIUNITE S.P.A. [IT/IT]; Viale Shakespeare, 47 I-00144 Roma (IT)
Launched – 1993 ITALY, SIGMA TAU, Non-Opioid Analgesics FOR Treatment of Osteoarthritis, Treatment of Rheumatoid Arthritis,
Originator sigma-tau SpA
Class Amino acids; Antipyretics; Nonsteroidal anti-inflammatories; Pyrroles; Small molecules
Mechanism of Action Cyclooxygenase inhibitors
Marketed Inflammation
Most Recent Events
01 Jun 1999 A meta-analysis has been added to the adverse events section
22 Jul 1995 Launched for Inflammation in Italy (PO)
Amtolmetin guacil is a NSAID which is a prodrug of tolmetin sodium.
Amtolmetin guacil is a nonacidic prodrug of tolmetin that has similar nonsteroidal antiinflammatory drug (NSAID) properties to those of Tolmetin with additional gastroprotective advantages. The term “nonsteroidal” is used to distinguish these drugs from steroids that have similar eicosanoid-depressing and antiinflammatory actions. Moreover, it possesses a more potent and long-lasting antiinflammatory activity than tolmetin and is marketed for the treatment of rheumatoid arthritis, osteoarthritis, and juvenile rheumatoid arthritis.
Background
Tolmetin sodium is an effective NSAID approved and marketed for the treatment of rheumatoid arthritis, osteoarthritis and juvenile rheumatoid arthritis. In humans, tolmetin sodium is absorbed rapidly with peak plasma levels observed 30 min after p.o. administration, but it is also eliminated rapidly with a mean plasma elimination t½ of approximately 1 hr. The preparation of slow release formulations or chemical modification of NSAIDs to form prodrugs has been suggested as a method to reduce the gastrotoxicity of these agents.
Amtolmetin guacil is a non-acidic prodrug of tolmetin, having similar NSAID properties like tolmetin with additional analgesic, antipyretic, and gastro protective properties. Amtolmetin is formed by amidation of tolmetin by glycine
Pharmacology
Almost is absorbed on oral administration. It is concentrated maximum in internal the gastric wall, and highest concentration reached in 2 hours after administration.
Amtolmetin guacil hydrolysed in to following metabolites Tolmetin, MED5 and Guiacol.
Elimination will complete in 24 hours. Happens mostly with urine in shape of gluconides products (77%), faecal (7.5%).
It is advised to take the drug on empty stomach.
Permanent anti-inflammatory action is continued up to 72 hours, with single administration.
Mechanism of action
Amtolmetin guacil stimulates capsaicin receptors present on gastro intestinal walls, because of presence of vanillic moiety and also releases NO which is gastro protective. It also inhibits prostaglandin synthesis and cyclooxygenase (COX).
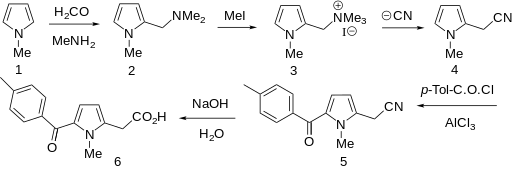
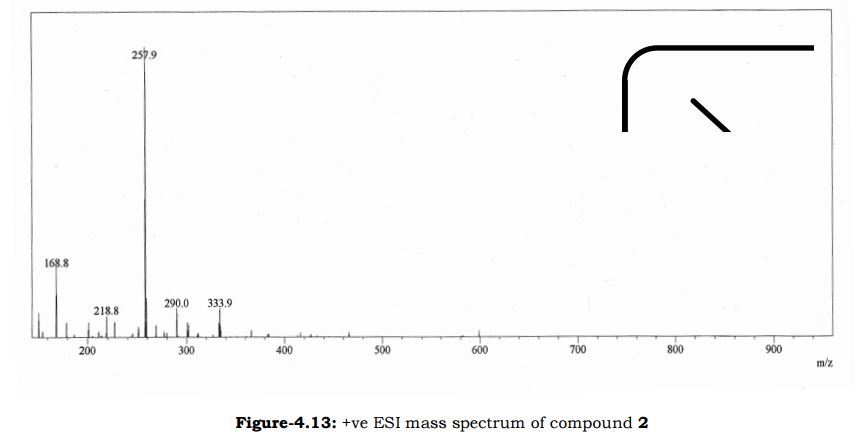
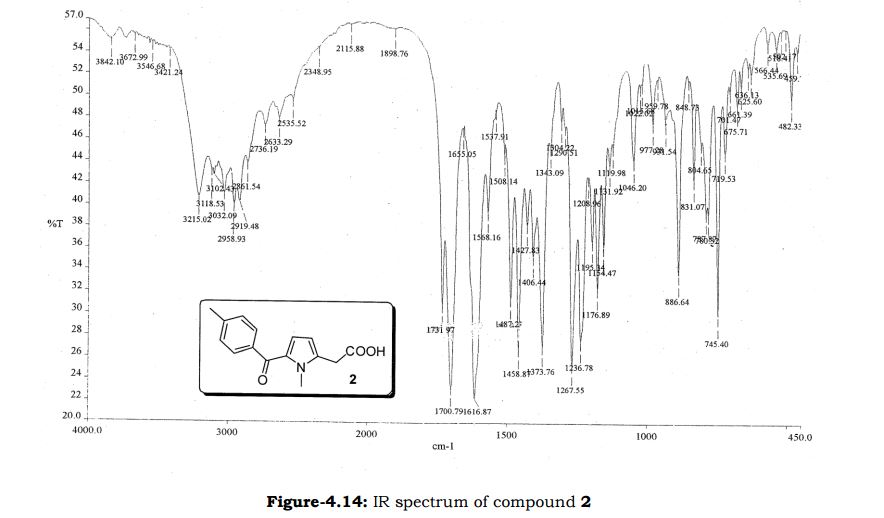
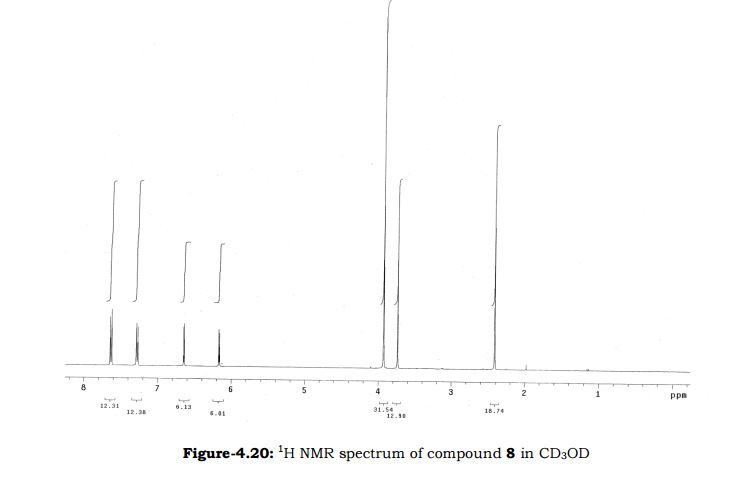
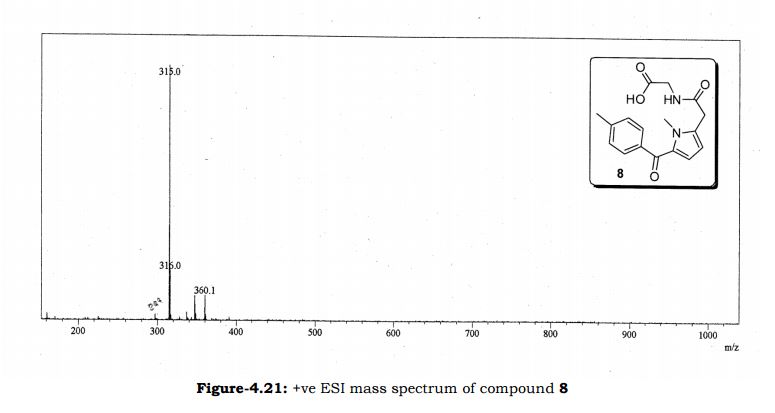
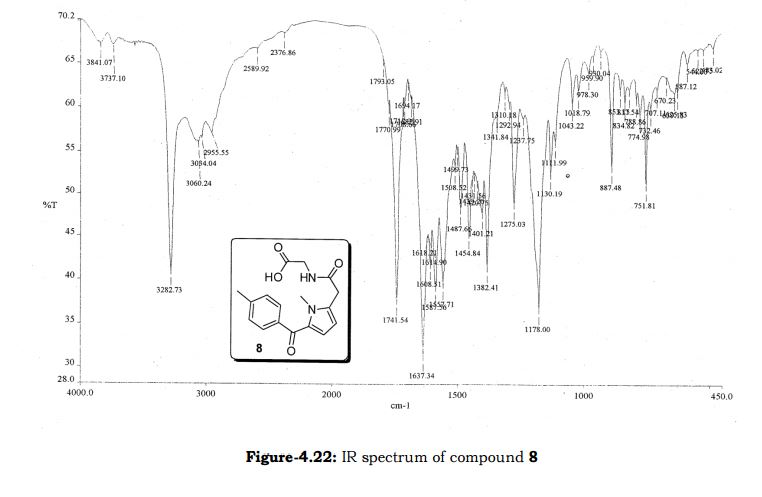
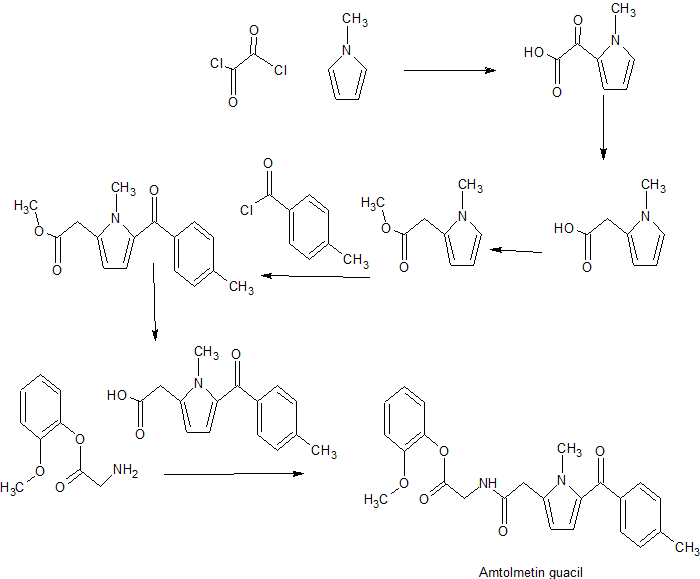
Amtolmetin guacil (CAS NO.: 87344-06-7), with its systematic name of N-((1-Methyl-5-p-toluoylpyrrol-2-yl)acetyl)glycine o-methoxyphenyl ester, could be produced through many synthetic methods.
Following is one of the synthesis routes: 1-Methyl-5-(4-methylbenzoyl)pyrrole-2-acetic acid (I) is condensed with glycine ethyl ester (II) in the presence of carbonyldiimidazole (CDI) and triethylamine in THF to afford the corresponding acetamidoacetate (III), which is hydrolyzed with NaOH in THF-water yielding 2-[2-[1-methyl-5-(4-methylbenzoyl)pyrrol-2-yl]acetamido]acetic acid (IV). Finally, this compound is esterified with 2-methoxyphenol (guayacol) (V) by means of CDI in hot THF.
The present invention relates to a new crystalline form of 1- methyl-5-p-toluoylpyrrole-2-acetamidoacetic acid guaiacyl ester, a process for its preparation and to pharmaceutical compositions endowed with antiinflammatory, analgesic and antipyretic activity containing same.
The ester of 1-methyl-5-p-toluoylpyrrole-2-acetamidoacetic acid (hereinafter referred to as MED 15, form 1) is a known compound.
In fact, US Patent 4,882,349 discloses a class of N-mono- substituted and N,N-disubstituted amides of l-methyl-5-p- toluoylpyrrole-2-acetic acid (known as Tolmetin) endowed of anti- inflammatory, analgesic, antipyretic, antisecretive and antitussive properties.
US Patent 4,578,481 claims a specific compound, endowed with valuable pharmacological activity, encompassed in the above- mentioned class, precisely 1-methyl-5-p-toluoylpyrrole-2-acetamido- acetic acid guaiacyl ester (which is MED 15, form 1), and a process for its preparation.
The process disclosed in US 4,578,481 presents some drawbacks, since it is not easily applicable on industrial scale and gives low yields.
According to the above-mentioned process, Tolmetin was reacted with N,N’-carbonyldiimidazole in tetrahydrofuran (THF), and aminoacetic acid ethyl ester hydrochloride was added to the reaction mixture.
Following a complex series of washings in order to remove the unreacted starting compounds, and crystallisation from benzene/ cyclohexane, 1-methyl-5-p-toluoylpyrrole-2-acetamidoace-tic acid ethyl ester was obtained. This compound was subsequently transformed into the corresponding acid.
The acid was reacted with N,N’-carbonyldiimidazole obtaining the corresponding imidazolide, to which a solution of guaiacol in
THF was added.
From the reaction mixture, following several washings, neutralisation and crystallisation from benzene/ cyclohexane MED 15 form 1 was obtained.
The main physico-chemical characteristics of MED 15 form 1 are shown in table 1, left column.
Image may be NSFW. Clik here to view.
Image may be NSFW. Clik here to view.
The above mentioned process comprises the following steps:
(a) hydrolysing TOLMETIN 1 methyl ester with an alkaline hydroxide in a basic environment, obtaining TOLMETIN 2 alkaline salt;
(b) condensing 2 with isobutylchloroformate 3 obtaining the mixed anhydride 4;
(d) condensing 6 with isobutylchloroformate 3 obtaining the mixed anhydride 7; and
(e) reacting the mixed anhydride 7 with guaiacol 8 obtaining 9 , MED 15, form 2.
The following non-limiting example illustrates the preparation of MED 15, form 2, according to the process of the present invention.
Preparation of 1-methyl-p-toluoylpirrol-2-acetoammidoacetic acid.
A mixture of 500 mL of toluene, 100 g of Tolmetin ethyl ester and 10 g of Terre deco in 1L flask, was heated to 70° C and maintained at this temperature for 20-30 min, under stirring. The mixture was then filtered on pre-heated buckner, and the solid phase washed with 50 mL of heated toluene. The discoloured toluene solution was transferred in a 2 L flask, 15 g of sodium hydroxide (97%) dissolved in 100 mL of water were added thereto.
The solution was heated at reflux temperature and refluxed for 1 hour. 22 mL of isobutyl alcohol were added to the solution which was heated at reflux temperature; water (about 120 mL) was removed completely with Marcusson’s apparatus arriving up to 104-105°C inner temperature.
To a suspension of Tolmetin sodium, cooled under nitrogen atmosphere to -5°C ± 2°C, 0.2 mL of N-methyl Morpholine were added. Maintaining the temperature at 0°C ± 3°C, 53 mL of isobutyl chloroformate were added dropwise in 5-10 min. After about 1 hour the suspension became fluid. Following 3 hours of reaction at 0°C + 3°C, over the glycine solution previously prepared, the mixed anhydride solution was added dropwise. The glycine solution was prepared in a flask containing 230 mL of demineralised water, 47 g of potassium hydrate (90%), cooling the solution to 20°C ± 2, adding 60 g of glycine, and again cooling to 10°C ± 2°C.
To the glycine solution, the mixed anhydride was added dropwise under stirring, in 5-10 min., maintaining the temperature at 20°C ± 2°C.
At the end of the addition, temperature was left to rise to room temperature, 1 hour later the reaction was complete. To the mixture 325 mL of demineralised water were added, the mixture was brought to pH 6.0 +2 using diluted (16%) hydrochloric acid (about 100 mL).
The temperature of the solution was brought to 73°C ±2°C and the pH adjusted to pH 5.0 ±0.2.
The separation of the two phases was made at hot temperature: the toluene phase was set aside for recovering acid-Tolmetin if any, the water phase was maintained at 73°C ±2°C and brought to pH 4.0 ±0.2 using diluted hydrochloric acid.
At the beginning of the precipitation the solution was slowly brought to pH 3.0 ±0.2 using diluted (16%) hydrochloric acid (100 mL).
The mixture was cooled to 15°C ±3°C and after 30 min. filtered. The solid cake was washed with 2×100 mL of demineralised water, the product was dried at 60°C under vacuum till constant weight. 100 g of 1-methyl-p-toluoylpirrol-2-acetoammidoacetic acid were obtained.
Preparation of MED 15, form 2
To a 2 L flask containing 730 mL of toluene, 100 g of dried compound of the above step were dissolved. To this solution 18.8 g of potassium hydrate (tit. 90%) in 65 mL of water were added.
The solution was dried maintaining the internal temperature at 95-100°C, and cooled to 55-60 °C. A solution of potassium hydrogen carbonate was then added and the resulting mixture was dried maintaining the internal temperature at 105°C ±2°C.
The mixture was cooled under nitrogen atmosphere to 5°C
±2°C, 24 mL of isobutyl alcohol and 0.3 mL of N-methyl morpholine were added thereto.
Maintaining the temperature at 10°C ±3°C, 47 mL of isobutyl-chloroformate were added dropwise in 5-10 minutes. The mixture was left to react for two hours at 10°C ±3°C obtaining an anhydride solution, which was added to a guaiacol solution previously prepared.
The guaiacol solution was prepared by loading in a 2L-flask 295 mL of water, 25 g of potassium hydrate (90%), and 0.3 g of sodium metabisulfite.
At the end of the loading the temperature was brought to 35-40°C.
The anhydride was added dropwise in 5- 10 min and the temperature was left to rise to room temperature.
The suspension was kept under stirring for 1 hour and brought to pH 6.0 ±0.5 with diluted hydrochloric acid. The suspension was heated to 70°C ± 5°C and maintained at pH 3-4 with diluted hydrochloric acid.
The phases were separated while hot. The aqueous phase was discharged, and to the organic phase, 250 mL of water were added.
Maintaining the temperature at 70 ±5°C the solution was brought to pH 8.0 ±0.5 with diluted sodium hydrate, the phases were separated while hot and the acqueous phase was discharged.
The organic phase was washed with 250 mL of water. At 70 ± 5°C the phases were separated. The toluene phase was then cleared with dicalite, filtered and left to crystallise.
The mixture was slowly cooled to 30°C – 35°C, the temperature was then brought to 10 ± 3°C and after 1 hour filtered, washed with toluene (2×100 mL).
The product was brought to dryness at 60°C under vacuum, thus giving 100 g of compound MED 15, form 2.
Equipped with a trap, 2000ml four-neck reaction flask with a mechanical stirrer and a thermometer, 加入托 US buna 100.0g (0.358mol) and 500ml of toluene, turned stirred and heated under reflux with toluene with water, drying the solution, when When the internal temperature reaches 95-100 ℃, the solution was cooled to 55-60 ℃, dissolved in 30ml of water was added portionwise 11.5g of potassium bicarbonate was added, and refluxed to remove water, until the internal temperature reaches 105 ± 2 ℃.The mixture was cooled to ice-water bath 5 ± 2 ℃, to which was added 24ml of isobutyl alcohol and 0.3ml N- methylmorpholine.The temperature was maintained at 10 ± 3 ℃, with a pressure-equalizing dropping funnel was added dropwise isobutylchloroformate 45.5ml (0.400mol), 10min addition was complete, so the mixture was 10 ± 3 ℃ 2hr reaction solution to obtain an acid anhydride, it has been prepared dropwise glycine guaiacol ester solution, 5-10min the addition was complete.Glycine guaiacol ester solution was prepared by adding 295ml of water in a 2000ml flask, 27g of potassium hydroxide (82%) and 0.3g of sodium metabisulfite, stirring to dissolve, the temperature was controlled at 10 ± 3 ℃, to which was added 82.7g (0.38mol) glycine guaiacol ester hydrochloride and prepared.Dropwise addition, the temperature was raised to room temperature, the reaction 2hr, diluted with 16% hydrochloric acid to adjust the mixture to pH 6.0 ± 0.5.The suspension was heated to 70 ± 5 ℃, and then 16% diluted hydrochloric acid to adjust the pH to 3.5 to 4.5, while hot liquid separation, discarding the aqueous phase, the organic phase was added to 250ml of water, maintaining the temperature at 70 ± 5 ℃ with dilute (2N) sodium hydroxide solution to adjust the solution to pH 8.0 ± 0.5, and then hot liquid separation, aqueous phase was discarded.With 2 × 250ml The organic phase was washed with water, the phases were separated at 70 ± 5 ℃, then clean the toluene organic phase through celite, cooled to room temperature, allowed to set freezer cooling crystallization, filtration, filter cake washed with 2 × 50ml of cold washed with toluene, and dried in vacuo at 60 ℃ to constant weight to give 1- methyl-5-p-toluoylpyrrole-2-acetamido acid guaiacol ester crude 135.5 g, yield 90%.The crude product was recrystallized from acetone to give 1-methyl-5-acyl-2-acetyl-p-toluene amino acid ester of guaiacol boutique 127.9 g, yield 94.4%, mp128.7 ~ 131.9 ℃.Elemental analysis: C, 68.53%; H, 5.76%; N, 6.65%.IR spectrum (KBr tablet method): 3318,3142,2963,1778,1652,1626,1605,1500,1480,1456,13731255 and 1153cm-1.
Example 2: Procedure: equipped with a water separator, 2000ml four-neck reaction flask with a mechanical stirrer and a thermometer, 加入托 US buna 100.0g (0.358mol) and 500ml of toluene, turned stirred and heated under reflux with toluene with water , drying the solution, when the internal temperature reaches 95-100 ℃, the solution was cooled to 55-60 ℃, dissolved in 30ml of water was added portionwise 11.5g of potassium bicarbonate was added, and refluxed to remove water, until the internal temperature reaches 105 ± 2 ℃.The mixture was cooled to ice-water bath 5 ± 2 ℃, to which was added 24ml of isobutyl alcohol and 0.3ml N- methylmorpholine.The temperature was maintained at 10 ± 3 ℃, with a pressure-equalizing dropping funnel dropwise isopropyl 46.5ml (0.41mol), 10-15min addition was complete, the mixture was allowed at 10 ± 3 ℃ reaction 2hr derived anhydride solution, it would have been prepared dropwise to glycine guaiacol ester solution, 5-10min the addition was complete.Glycine guaiacol ester solution was prepared by adding 295ml of water in a 2000ml flask, 27g of potassium hydroxide (82%) and 0.3g of sodium metabisulfite, stirring to dissolve, the temperature was controlled at 10 ± 3 ℃, to which was added 82.7g (0.38mol) glycine guaiacol ester hydrochloride and prepared.Dropwise addition, the temperature was raised to room temperature, the reaction 2hr, diluted with 16% hydrochloric acid to adjust the mixture to pH 6.0 ± 0.5.The suspension was heated to 70 ± 5 ℃, and then 16% diluted hydrochloric acid to adjust the pH to 3.5 to 4.5, while hot liquid separation, discarding the aqueous phase, the organic phase was added to 250ml of water, maintaining the temperature at 70 ± 5 ℃ with dilute (2N) sodium hydroxide solution to adjust the solution to pH 8.0 ± 0.5, and then hot liquid separation, aqueous phase was discarded.With 2 × 250ml The organic phase was washed with water, the phases were separated at 70 ± 5 ℃, then clean the toluene organic phase through celite, cooled to room temperature, allowed to set freezer cooling crystallization, filtration, filter cake washed with 2 × 50ml of cold washed with toluene, and dried in vacuo at 60 ℃ to constant weight to give 1- methyl-5-p-toluoylpyrrole-2-acetamido acid guaiacol ester crude 138.5 g, yield 92%.The crude product was recrystallized from acetone to give 1-methyl-2-acyl-5-toluene acetaminophen acid ester guaiacol boutique 128.8 grams.
Example 3: equipped trap, 2000ml four-neck reaction flask with a mechanical stirrer and a thermometer, 加入托 US buna 100.0g (0.358mol) and 500ml of toluene, turned stirred and heated under reflux with toluene with water, dried solution, when the internal temperature reaches 95-100 ℃, the solution was cooled to 55-60 ℃, dissolved in 30ml of water was added portionwise 10-12.5g potassium bicarbonate solution, refluxing was continued for removal of water, until the internal temperature reaches 105 ± 2 ℃.The mixture was cooled to ice-water bath 5 ± 2 ℃, added thereto 20-30ml of isobutyl alcohol 0.2-0.5mlN- methylmorpholine.The temperature was maintained at 10 ± 3 ℃, with a pressure-equalizing dropping funnel was added dropwise isobutylchloroformate 40.5-48.5ml, 10-15min addition was complete, so the mixture was 10 ± 3 ℃ 2hr reaction solution to obtain an acid anhydride, it has been prepared dropwise glycine guaiacol ester solution, 5-10min the addition was complete.Glycine guaiacol ester solution was prepared by adding 295ml of water in a 2000ml flask, 25-30g of potassium hydroxide (82%) or 15-17 grams of sodium hydroxide and sodium metabisulfite 0.2-0.5g or insurance powder, stirring to dissolve the temperature is controlled at 10 ± 3 ℃, to which is added 80-84g glycine guaiacol ester hydrochloride and prepared.Dropwise addition, the temperature was raised to room temperature, the reaction 2hr, the mixture was adjusted with dilute hydrochloric acid to pH 6.0 ± 0.5.The suspension was heated to 70 ± 5 ℃, with dilute hydrochloric acid to adjust the pH to 3.5 to 4.5, while hot liquid separation, discarding the aqueous phase, the organic phase was added to 250-280ml of water, maintaining the temperature at 70 ± 5 ℃ , adjusted with dilute sodium hydroxide solution and the solution to pH 8.0 ± 0.5, and then hot liquid separation, aqueous phase was discarded.With 2 × 250ml The organic phase was washed with water, the phases were separated at 70 ± 5 ℃, then clean the toluene organic phase through celite, cooled to room temperature, allowed to set freezer cooling crystallization, filtration, filter cake washed with 2 × 50ml of cold washed with toluene, and dried in vacuo at 60 ℃ to constant weight to give 1- methyl-5-p-toluoylpyrrole-2-acetamido acid guaiacol ester crude 130-139 grams.The crude product was recrystallized from acetone to give 1-methyl-5-acyl-2-acetyl-p-toluene amino acid ester boutique guaiacol 120-129 grams.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides safe, environment friendly, economically viable and commercially feasible processes for the production of Amtolmetin guacil. There are two methods for the preparation of Amtolmetin guacil. The processes for the production of Amtolmetin guacil (I) comprise:
Method-1:
Step-A:- Treating 2-methoxy phenol of Formula VI with 2-(benzyloxycarbonylamino) acetic acid of Formula VII in the presence of an organic base and a condensing agent in chlorinated solvent to yield 2-methoxyphenyl-2- (benzyloxycarbonylamino) acetate of Formula V.
Step-B:- Acid addition salt of 2-methoxyphenyl -2-aminoacetate of Formula II may be prepared by treating 2-methoxyphenyl-2- (benzyloxycarbonylamino) acetate of Formula V with an acid and followed by crystallization in aprotic solvent.
7
Step-C):- l-methyl-5-p-toluoylpyrrole-2-acetic acid of Formula III is reacted with a condensing agent to form-activated moiety, which is reacted with acid addition salt of 2-methoxyphenyl -2-aminoacetate of Formula II in chlorinated solvent to produce Arntolmetin guacil of formula (I).
In a preferred embodiment of present invention, condensing agent used in step-A is selected from group consisting of dicyclohexylcarbodiimide, N, N’-carbonyl diimidazole, hydroxy benzotriazole. The most preferred condensing agent is Dicyclohexyl carbodiimide for the reaction.
The solvent used in present invention is selected from the group consisting of but not limited to toluene, methylene chloride, chloroform, water miscible ethers such as tetrahydrofuran, 1,4-dioxane, the most preferred solvent for the reaction methylene dichloride.
In another embodiment of the present invention, the reaction is performed in the presence of an organic base. The organic base is selected from the group consisting of trimethylamine, triethylamine, N-methyl morpholine, N-methylpyrrolidinone, 4-dimethyl Aminopyridine; the most preferred base is 4-dimethyl Aminopyridine.
In a preferred embodiment of present invention, the non-polar solvent used in step-B is selected from group consisting of ethers, hexanes, aromatic hydrocarbons and esters.
In another preferred embodiment of present invention, the most suitable solvents are esters.
In another preferred embodiment of present invention, condensing agent used in step-C is selected from group consisting of dicyclohexylcarbodiimide, N, N’-carbonyl diimidazole, hydroxy benzotriazole. The most preferred condensing agent is N, N’-carbonyl diimidazole for the conversion of the reaction.
8
The solvent used in present invention is selected from the group consisting of but not limited to toluene, methylene chloride, chloroform, water miscible ethers such as tetrahydrofuran, 1,4-dioxane, the most preferred solvent for the reaction methylene dichloride.
In yet another embodiment of the present invention, the reaction is performed at a temperature in the range of -20°C to 50°C. Most preferred temperature range for the reaction is (-) 10°C to 0°C.
Method-2:
Treating 2-(2-(I-methyl-5- (4-methylbenzoyl)-lH-pyrrol-2-yl) acetamido) acetic acid with 2-methoxy phenol in presence of condensing reagent and an organic base to obtain Amtolmetin guacil.
In a preferred embodiment of present invention, the condensing agent used is selected from group consisting of dicyclohexyicarbodiimide, hydroxy benzotriazole or a mixture thereof. The most preferred condensing agent is Dicvclohexyl carbodiimide for the aforementioned reaction.
The solvent used in present invention is selected from the group consisting of but not limited to toluene, methylene chloride, chloroform, water miscible ethers such as tetrahydrofuran. 1,4-dioxane, the most preferred solvent for the reaction is methylene dichloride.
In another embodiment of the present invention, the reaction is performed in the presence of an organic base. The organic base is selected from the group consisting of triethylamine, triethylamine, N-methyl morpholine, N-methylpyrrolidinone, 4-dimethyl Aminopyridine; the most preferred base is 4-dimethyl Aminopyridine.
9
In yet another embodiment of the present invention, the reaction is performed at a temperature in the range of -20°C to 50°C. Most preferred temperature range for the reaction is (-) 10°C to 0°C.
In another embodiment of present invention, crude amtolmetin guacil is directly purified using polar and non-polar solvent or a mixture thereof. The most preferred solvents are Isopropanol and toluene.
The following non-limiting examples illustrate specific embodiments of the present invention. They are, however, not intended to be limiting the scope of present invention in anyway.
Preparation of Amtolmetin guacil: Example-1;
Charged MDC (600 ml) and N-benzyloxycarbonyl glycine (100 gm) in a 2L-4NRBF under N2 atmosphere. Reaction mass was cooled down to -5°C. Added N, N’-dicyclohexylcarbodiimide solution (108.5 gm in 300 ml MDC) at-5°C to 0°C. Maintained temperature of reaction for 10 minutes at -5°C to 0°C. Added guaiacol solution (59.36 gm in 180 ml MDC) at -5°C to 0°C followed by addition of N, N-dimethyl aminopyridine (1 gm) at -5°C to 0°C. Monitored the reaction over TLC till the completion of reaction, while maintaining reaction at 0°C. Filtered the undissolved Dicyclohexyl urea and washed the solids with methylene dichloride (125 ml X 2). Collected filtrate and washing. Washed methylene dichloride with water (1000 ml X 2), lN-NaOH (500 ml X 2) and 1% HC1 solution (500 ml X 2), water (500 ml X 2) respectively. Organic methylene dichloride layer was dried over anhydrous sodium sulphate. Filtered sodium sulphate and collected methylene dichloride filtrate. Distilled out methylene dichloride under vacuum below 40°C to get oil. HPLC purity :> 90%
10
Added 33% HBr in acetic acid solution (262,5 gm) into reaction vessel at 25-30°C. Monitored the reaction over TLC till the completion of reaction, while maintaining the reaction at 25-30°C. Added ethyl acetate (1200 ml) slowly at 25-30°C after completion of reaction. Stirred the resultant slurry for 2.5 hours at 25-30°C for complete crystallization. Filtered the solids and washed it with ethyl acetate (200 ml). Dried solids at 50-55°C. Dry weight: 102 gm. HPLC Purity: >98%
Example-2:
Charged MDC (1400 ml) and N, N’-carbonyl di imidazole (69.34 gm) into a 3L-4NRBF under N2 atmosphere. Cooled it down to -15°C. Charged Tolmetin acid (100 gm) slowly into reaction vessel at -10° ± 5°C. Monitored the progress of reaction of over HPLC. After completion of reaction, charged slowly 2-methoxyphenyl-2- (benzyloxy carbonylamino) acetate hydrobromide salt (112.05 gm) at -10° ± 5°C.Monitored the reaction over HPLC. After completion of reaction, washed the organic layer with water (300 ml), 1% NaOH solution (100 ml) and water (300 ml X 2) respectively at 3-8°C. Treated organic layer with activated carbon (2.5 gm) and filtered over hyflow bed. Washed hyflow bed with methylene dichlonde (100 ml X 2). Distilled out methylene dichloride below 40°C under vacuum and stripped off traces with toluene (100 ml X 2) at 50-55°C. Charged toluene (600 ml) and Isopropanol (50ml). Heated the mass to 63-68°C. Stirred the clear solution at 63-68°C for 1 hour. Cooled it down slowly to 30°C followed by further cooling to 5°C. Stirred the resultant slurry for 3 hours at 0-5°C. Filtered solids and washed with toluene (100 ml X 2). Dried solids at 55-60°C under vacuum. Dry Weight: 130 gm. HPLC Purity: >99%
Example-3:
Charged MDC (333 liter) and 2-(2-(l-methyl-5- (4-methylbenzoyl)-lH-pyrrol-2-yl) acetamido) acetic acid (55.5 Kg) in reactor under N2 atmosphere at 25-30°C. Cool down reaction mass to -15 to -12°C. Added a freshly prepared solution of N, N’-dicyclohexyl
11
carbodiimide (47.39 Kg in 166.5 liter) slowly at -10° ± 5°C within 1 hour. Rinsed the addition funnel with MDC (55.5 liter) and added it to the reaction at -10° ± 5°C. Added guaiacol solution (24.14 Kg in 99.9 liter MDC) to the reaction mass at -10° ± 5°C within 1 hour. Rinsed the addition funnel with MDC (11.1 liter) and added to the reaction -10° ± 5°C. Charged N, N’-dimethyl aminopyridine (0.555 Kg) at -15°C. Maintained temperature of reaction mass at -10° ± 5°C for 3 hours. Monitored the reaction over TLC, After the completion of reaction, filtered the dicyclohexyl urea and washed the solids with MDC (55.5L X 2). Collected MDC filtrate and wash it with water (166.5 L X 2). Collected MDC layer and treated it with activated carbon (2.77 Kg) and filtered through sparkler. Washed the sparkler with MDC (111 L). Distilled out MDC below 40°C under vacuum and stripped off traces with toluene (55.5 L X 2) at 50-55°C. Charge toluene (333L) and Isopropanol (27.75 L). Heated reaction mass to 63-68°C to get a clear solution. Stirred the clear solution at 63-68°C for 1 hour. Cooled it down slowly to 30°C followed by further cooling to 20oC. Stirred the resultant slurry for 2 hours at 17-20°C. Filtered the solids and washed with toluene (55.5 L X 3). Dried the solids at 55-60°C under vacuum. Dry Weight: 48 Kg. HPLC Purity:>99%
PAPER
Synthesis and Process Optimization of Amtolmetin: An Antiinflammatory Agent†
Center of Excellence, Integrated Product Development, Innovation Plaza, Dr. Reddy’s Laboratories Ltd., Bachupalli, Qutubullapur, R. R. Dist. 500 072 Andhra Pradesh, India, and Center for Environment, Institute of Science and Technology, Jawaharlal Nehru Technological University, Kukatpally, Hyderabad 500 072, India
Efforts toward the synthesis and process optimization of amtolmetin guacil 1 are described. High-yielding electrophilic substitution followed by Wolf−Kishner reduction are the key features in the novel synthesis of tolmetin 2 which is an advanced intermediate of 1.
Percent Composition: C 68.56%, H 5.75%, N 6.66%, O 19.03%
Literature References: Ester prodrug of tolmetin, q.v. Prepn: A. Baglioni, BE896018; idem,US4578481 (1983, 1986 both to Sigma-Tau). Pharmacology: E. Arrigoni-Martelli, Drugs Exp. Clin. Res.16, 63 (1990); A. Caruso et al.,ibid.18, 481 (1992). HPLC determn in plasma: A. Mancinelli et al.,J. Chromatogr.553, 81 (1991). Series of articles on pharmacokinetics and clinical trials:Clin. Ter.142 (1 pt 2) 3-59 (1993).
Properties: Crystals from cyclohexane-benzene, mp 117-120°. Sol in common organic solvents. LD50 in male mice, rats (mg/kg): 1370, 1100 i.p.; >1500, 1450 orally (Baglioni).
Melting point: mp 117-120°
Toxicity data: LD50 in male mice, rats (mg/kg): 1370, 1100 i.p.; >1500, 1450 orally (Baglioni)
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Director at Dr. Reddy’s Laboratories, Vice-Chair, ACS-India Chapter (South India)
Image may be NSFW. Clik here to view.
They have developed an improved process for pioglitazone which appears to be more compatible with industrial scale and has some advantages over the existing synthesis.
Preparation of Pioglitazone Hydrochloride (1·HCl) salt
Innovation Plaza, IPD, R&D, Dr. Reddy’s Laboratories Ltd., Survey Nos. 42, 45,46, and 54, Bachupally, Qutubullapur, R.R. District – 500 073, Andhra Pradesh, India, and Institute of Science and Technology, Center for Environmental Science, JNT University, Kukatpally, Hyderabad – 500 072, Andhra Pradesh, India
Org. Process Res. Dev., 2009, 13 (6), pp 1190–1194
An improved process for pioglitazone (1) is described. The process features high-yielding transformations employing inexpensive reagents and recoverable solvents.
Mechanism of ActionCalcitonin gene-related peptide receptor antagonists
Migraine is a neurovascular disorder characterized by severe, debilitating, and throbbing unilateral headache. Though a leading cause of disability, it is a highly prevalent disease with a clear unmet medical need. With the significant progress achieved in the field of pathophysiology in the past decades, to date, it is well recognized that the neuropeptide calcitonin gene-related peptide (CGRP), which is expressed mainly in the central and peripheral nervous system, plays a crucial role in migraine. Antagonism of CGRP receptors, as a potential new therapy for the treatment of migraine, could offer the advantage of avoiding the cardiovascular liabilities associated with other existing antimigraine therapies.
The calcitonin gene-related peptide (CGRP) is a strong vasodilator primarily found in nervous tissue. Since vasodilation in the brain is thought to be involved in the development of migraine and CGRP levels are increased during migraine attacks, this peptide may be an important target for potential new antimigraine drugs.
A Phase IIa clinical trial studying telcagepant for the prophylaxis of episodic migraine was stopped on March 26, 2009 after the “identification of two patients with significant elevations in serum transaminases”.[4] A memo to study locations stated that telcagepant had preliminarily been reported to increase the hepatic liver enzyme alanine transaminase (ALT) levels in “11 out of 660 randomized (double-blinded) study participants.” All study participants were told to stop taking the medication.[5]
On July 29, 2011, it was reported that Merck & Co. were discontinuing the clinical development program for telcagepant. According to Merck, “[t]he decision is based on an assessment of data across the clinical program, including findings from a recently completed six-month Phase III study”.[6]
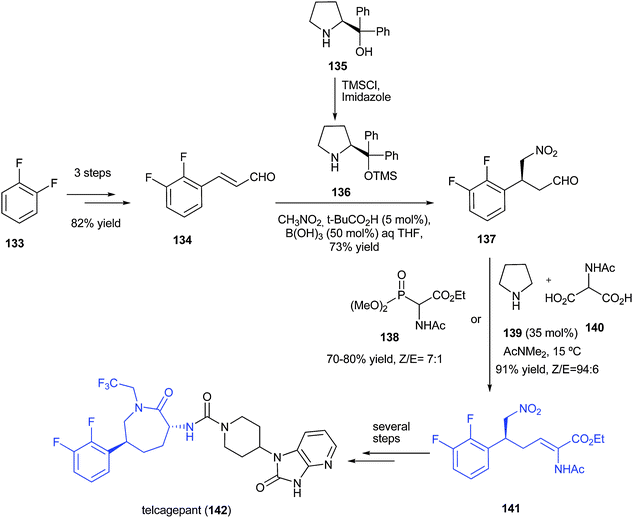
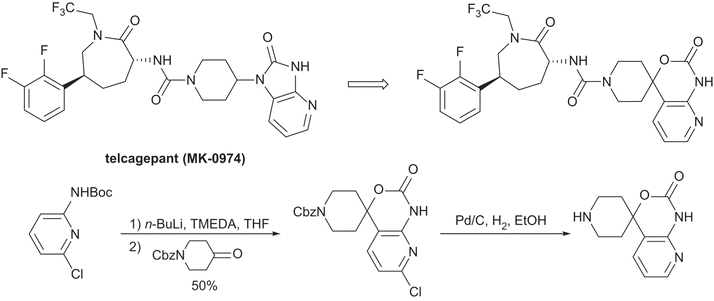
As part of the process of bringing a new API to market, it is often required to use an alternative synthetic strategy to the initial medicinal chemistry approach. Here Xu et al. of Merck Rahway disclose their efforts towards an improved multikilogram synthesis of telcagepant, a CGRP receptor antagonist for the treatment of migraines ( J. Org. Chem. 2010, 75, 7829−7841). The route described in the report is an example of a synthetic target driving the discovery of new chemistries.
Image may be NSFW. Clik here to view.
Of note are the challenges they faced and overcame in particular the asymmetric Michael addition of nitromethane to a cinnamyl aldehyde. Initial attempts under Hayashi’s conditions gave promising results (50−75% yield) and moreover confirmed a high enantioselectivity could be achieved using the Jorgensen−Hayashi catalyst. However, the use of benzoic acid as the acidic cocatalyst gave rise to undesired byproducts. After performing a comprehensive screen of conditions Xu showed that the combination of the weak acids t-BuCO2H (5 mol %) and B(OH)3(50 mol %) minimized the level of impurities. Of specific note is that this is the first reported application of iminium organocatalysis on industrial scale.
Image may be NSFW. Clik here to view.
The second milestone achieved in the strategy was the prevention of the protodefluorination under hydrogenative conditions. During the initial studies between 1.06−2.5% of the desfluoro compounds were formed by using Pd(OH)2/C in 100% conversion. To suppress the by product formation Xu screened a range of inorganic additives and found that 0.3 eq of LiCl gave a reproducible reaction where less than 0.2% of the desfluoro compounds were generated.
telcagepant as its crystalline potassium salt ethanol solvate in 92% yield with >99.9% purity and >99.9% ee.
Process for the Preparation of Pyridine Heterocycle Cgrp Antagonist Intermediate
2009-07-09
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
Hana and co-workers ( Synlett 2010, 18, 2759−2764) from Genentech have developed a single-step procedure for conversion of 2-nitro aromatic amines to benzimidazoles. Addition of ammonium chloride proved necessary as Fe powder and formic acid alone was ineffective for nitro reduction. These conditions were compatible with a variety of functional groups on the aromatic, including boronate esters. The methodology was also extended to nitro aminopyridines but failed to deliver the desired product with isoxazole or pyrazole reactants.
Mild and General One-Pot Reduction and Cyclization of Aromatic and Heteroaromatic 2-Nitroamines to Bicyclic 2H-Imidazoles
Emily J. Hanan*, Bryan K. Chan, Anthony A. Estrada, Daniel G. Shore, Joseph P. Lyssikatos
*Discovery Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, CA 94080, USA, Email: hanan.emilyImage may be NSFW. Clik here to view.gene.com
E. J. Hanan, B. K. Chan, A. A. Estrada, D. G. Shore, J. P. Lyssikatos, Synlett, 2010, 2759-2764.
A one-pot procedure for the conversion of aromatic and heteroaromatic 2-nitroamines into bicyclic 2H-benzimidazoles employs formic acid, iron powder, and NH4Cl as additive to reduce the nitro group and effect the imidazole cyclization with high-yielding conversions generally within one to two hours. The compatibility with a wide range of functional groups demonstrates the general utility of this procedure.
Image may be NSFW. Clik here to view.
see article for more examples
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
“ALL FOR DRUGS” CATERS TO EDUCATION GLOBALLY, No commercial exploits are done or advertisements added by me. This article is a compilation for educational purposes only.
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent
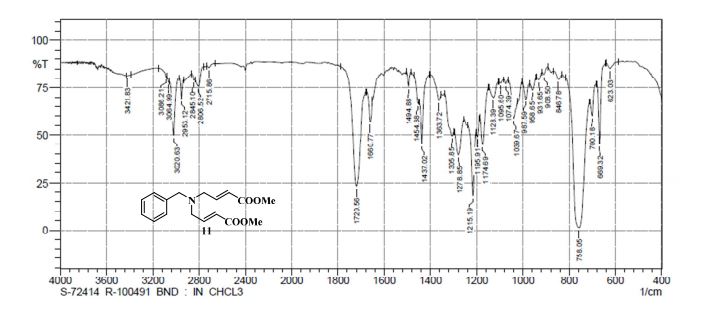
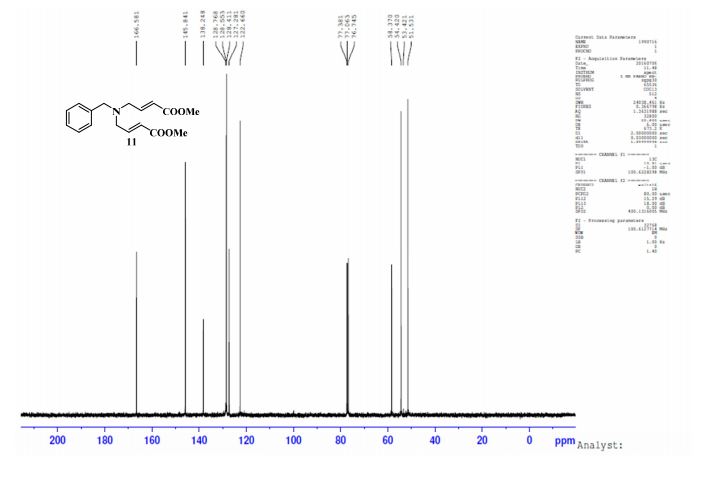
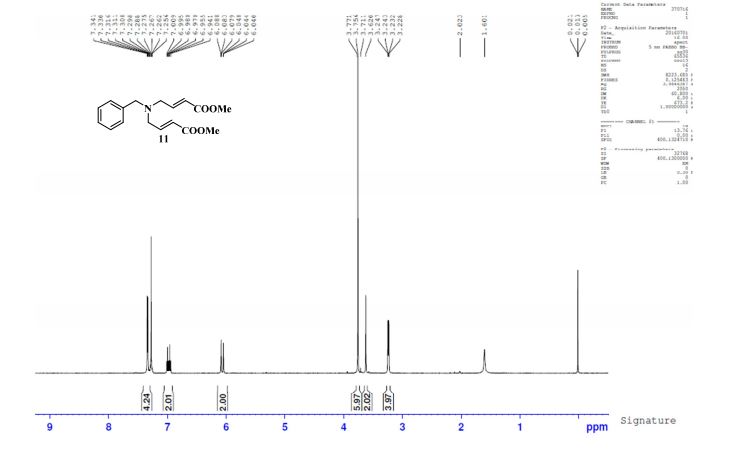
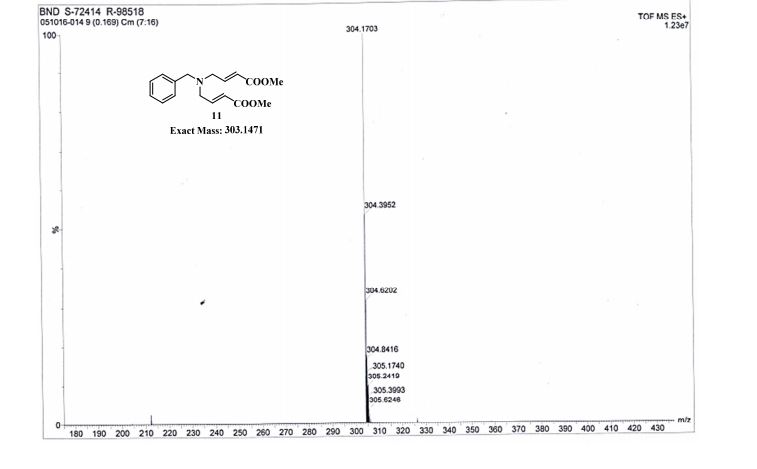
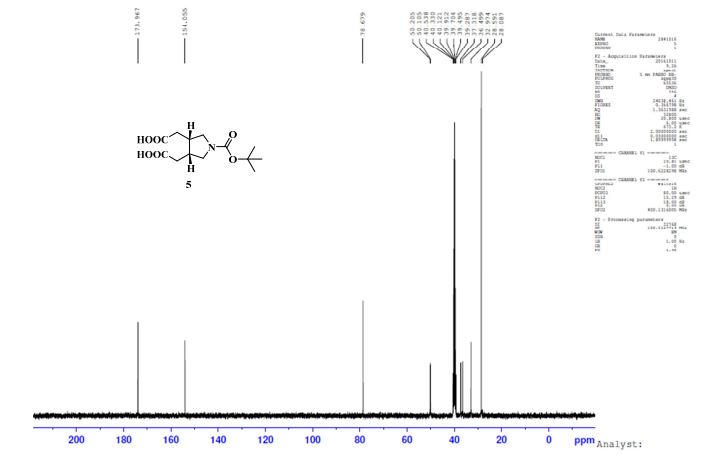
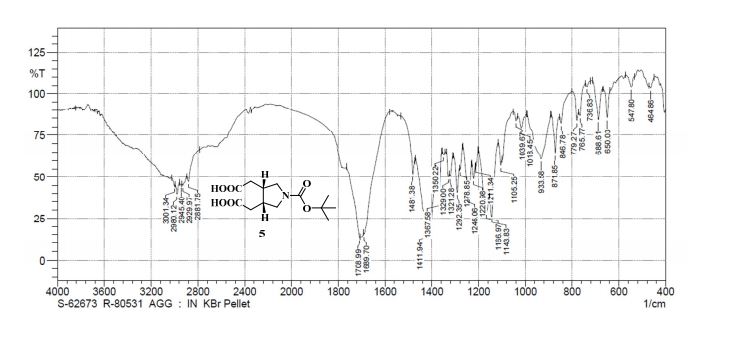
UPLC conditions were as follows for compound 11; Acquity Waters, column: BEH C18 (2.1 mm X 100 mm) 1.7 µm with mobile phases A (0.05% TFA in water) and B (acetonitrile). Detection was at 220 nm, flow was set at 0.4 mL/min, and the temperature was 30 °C (Run time: 9 min). Gradient: 0 min, A = 90%, B = 10%; 0.5 min, A = 90%, B = 10%; 6.0 min, A = 0%, B = 100%; 7.5 min, A = 0%, B = 100%; 7.6 min, A = 90%, B = 10%; 9.0 min, A = 90%, B = 10%.
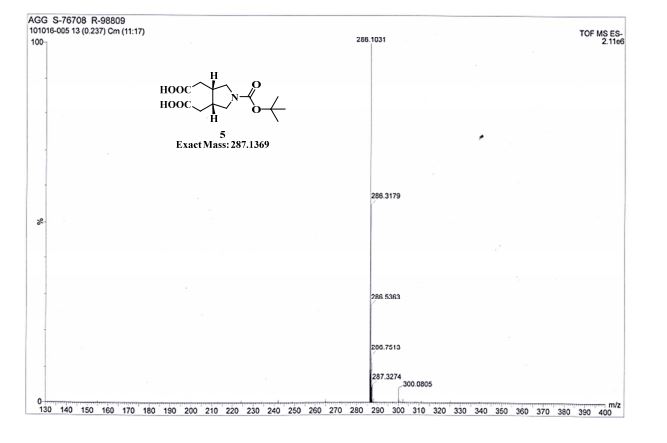
TOFMS: [C13H21NO6 – H+]: calculated 286.1296, found 286.1031(100%).
HPLC conditions were as follows for compound ; Agilent 1100 series, column: YMC J’SPHERE C18 (150 mm X 4.6 mm) 4µm with mobile phases A (0.05% TFA in water) and B (acetonitrile). Detection was at 210 nm, flow was set at 1.0 mL/min, and the temperature was 30 °C (Run time: 45 min). Gradient: 0 min, A = 90%, B = 10%; 5.0 min, A = 90%, B = 10%; 25 min, A = 0%, B = 100%; 30 min, A = 0%, B = 100%, 35 min, A = 90%, B = 10%; 45 min, A = 90%, B = 10%.
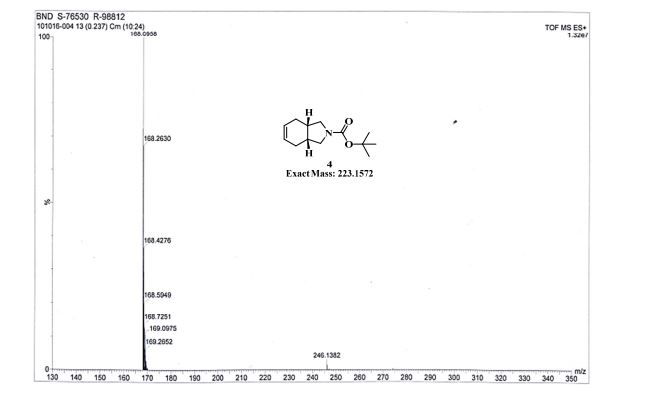
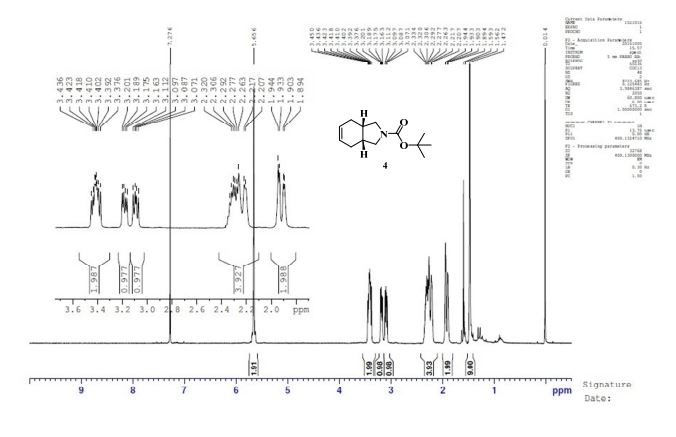
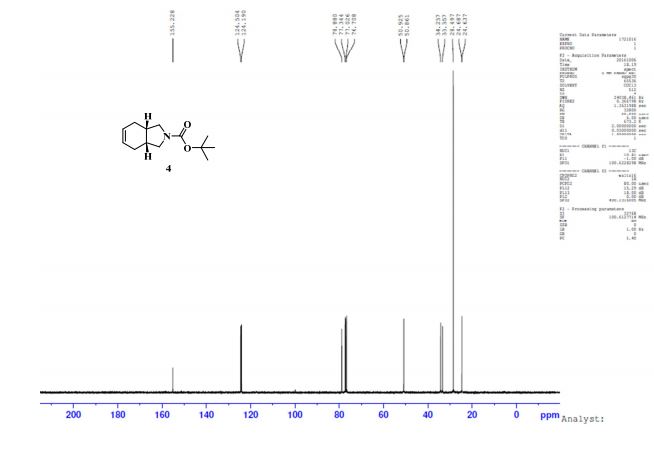
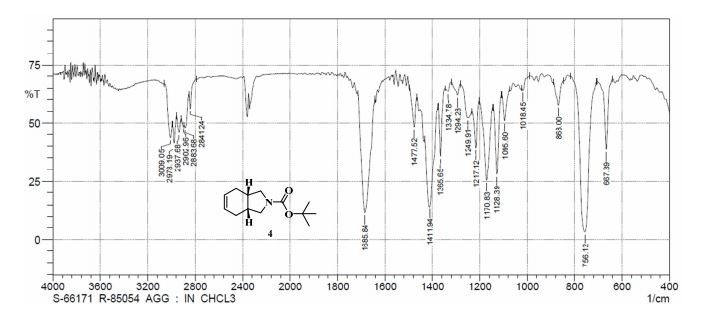
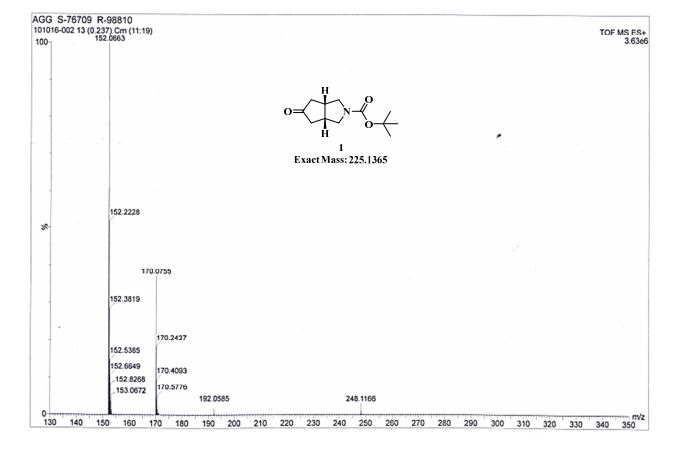
GC conditions were as follows for compound 4; Agilent GC-FID 7890A, column: ZB-5MSi (30 m X 0.32 mm, 0.25 µm) with injector temperature 250 ºC and detector temperature 280 ºC, diluent was Methanol, Oven temperature was at 70 ºC isocratic for 3 min. then raised up to 250 ºC @ 20 ºC/min then 15 min. hold.
Anal. Calcd for C12H19NO3: C, 63.98; H, 8.50; N, 6.22. Found: C, 63.89; H, 8.27; N, 5.97.
HPLC conditions were as follows for compound ; Agilent 1100 series, column: YMC J’SPHERE C18 (150 mm X 4.6 mm) 4µm with mobile phases A (0.05% TFA in water) and B (acetonitrile). Detection was at 210 nm, flow was set at 1.0 mL/min, and the temperature was 30 °C (Run time: 45 min). Gradient: 0 min, A = 90%, B = 10%; 5.0 min, A = 90%, B = 10%; 25 min, A = 0%, B = 100%; 30 min, A = 0%, B = 100%, 35 min, A = 90%, B = 10%; 45 min, A = 90%, B = 10%.
At award function for my award “100 Most Impactful Health care Leaders Global listing”, conferred on me at Taj lands end, Mumbai, India on 14 Feb 2014 by World Health Wellness congress and awards
 Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW. Image may be NSFW.
Image may be NSFW.
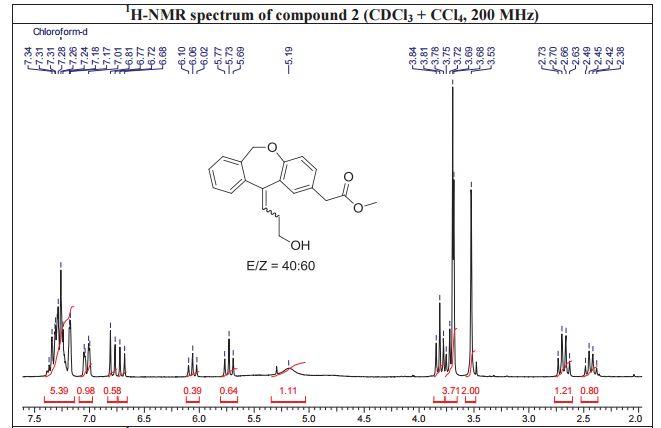
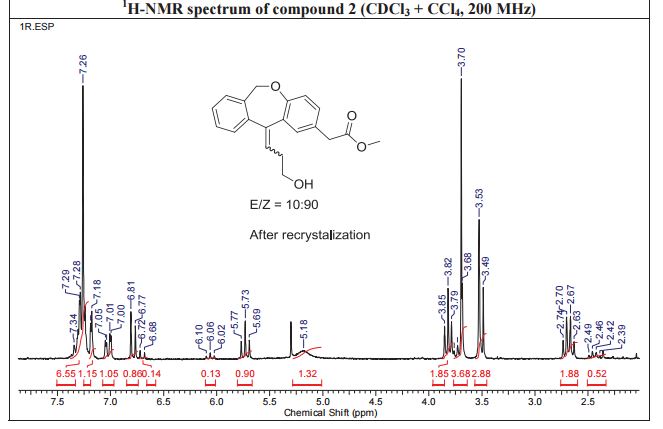
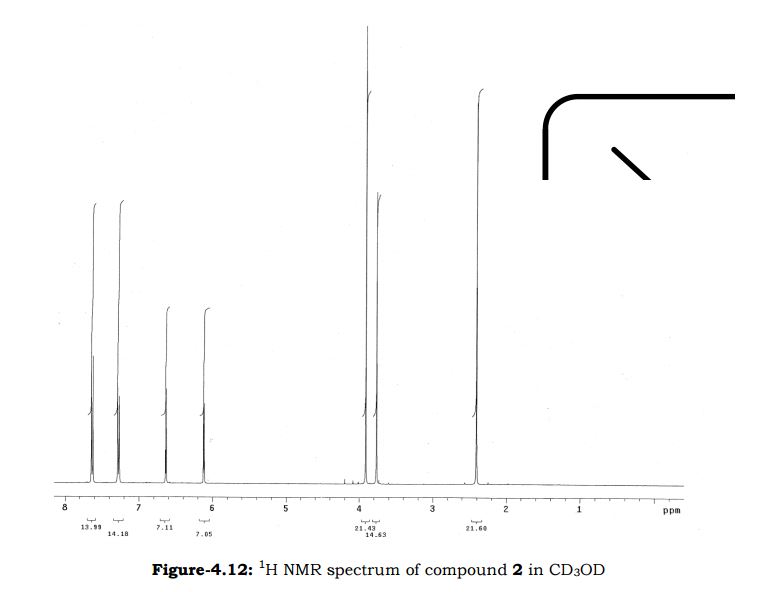
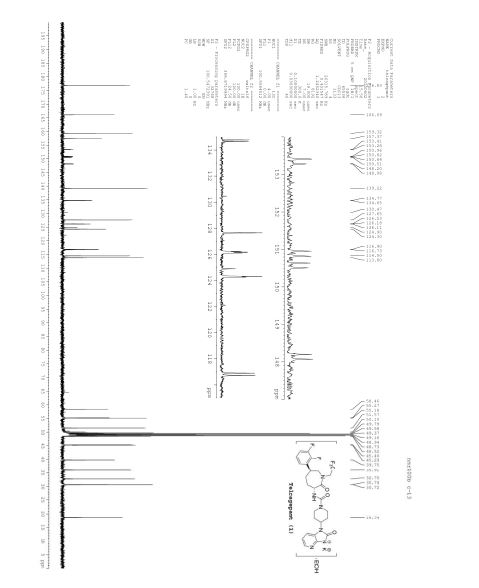
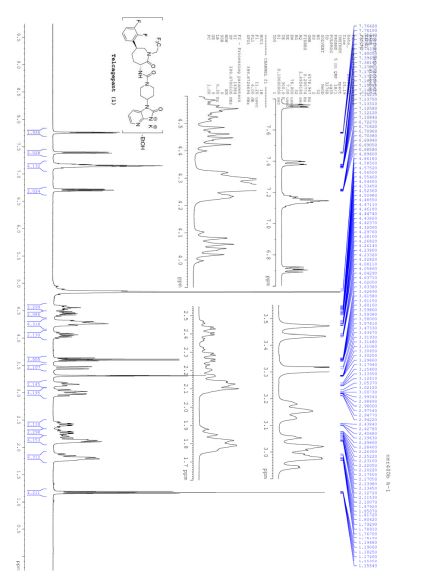
 NMR IS EASY
NMR IS EASY